
Phylogenomic and Microsynteny Analysis Provides Evidence of Genome Arrangements of High-Affinity Nitrate Transporter Gene Families of Plants

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
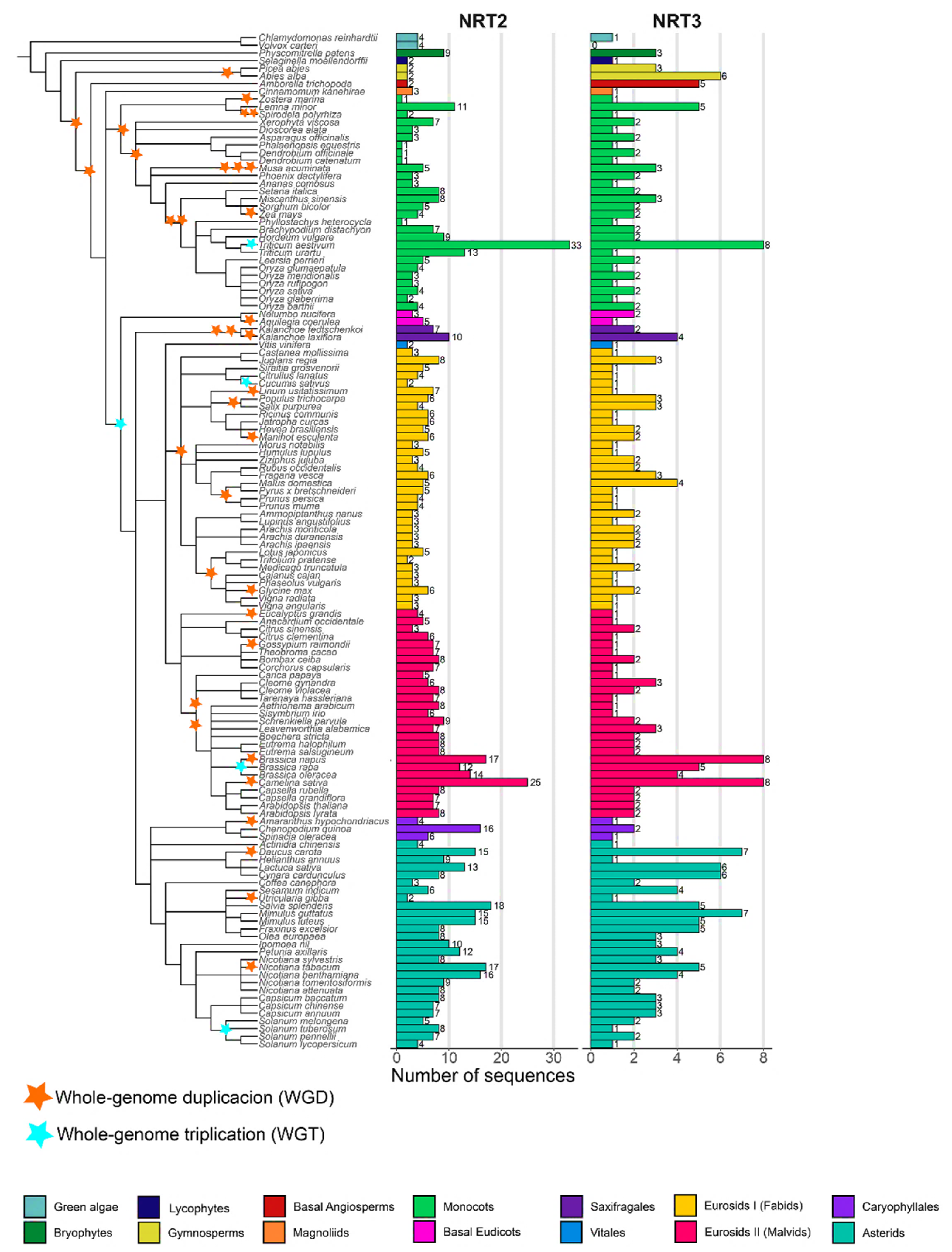
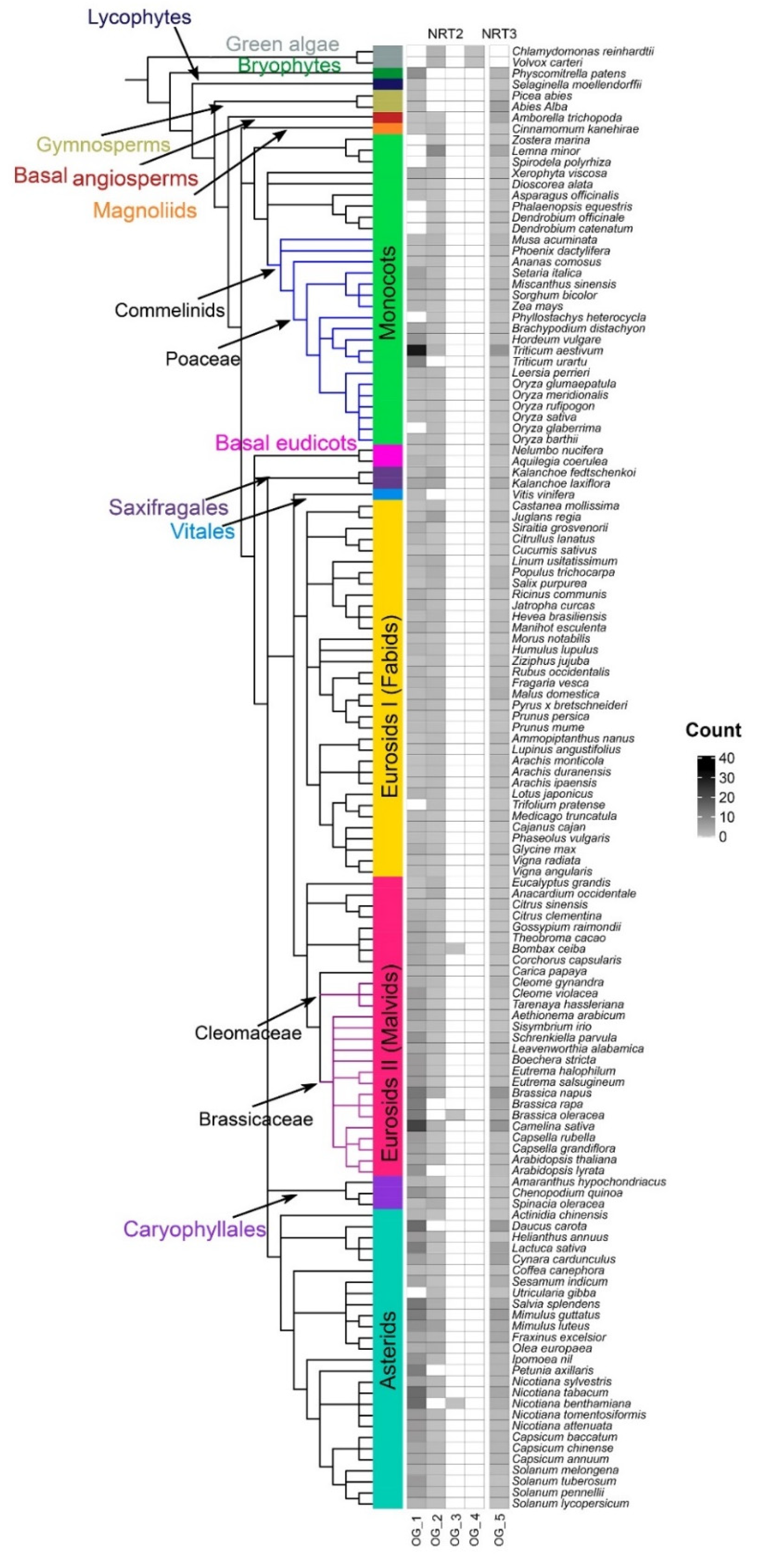
2.1. NRT2 and NRT3 Sequences Identification
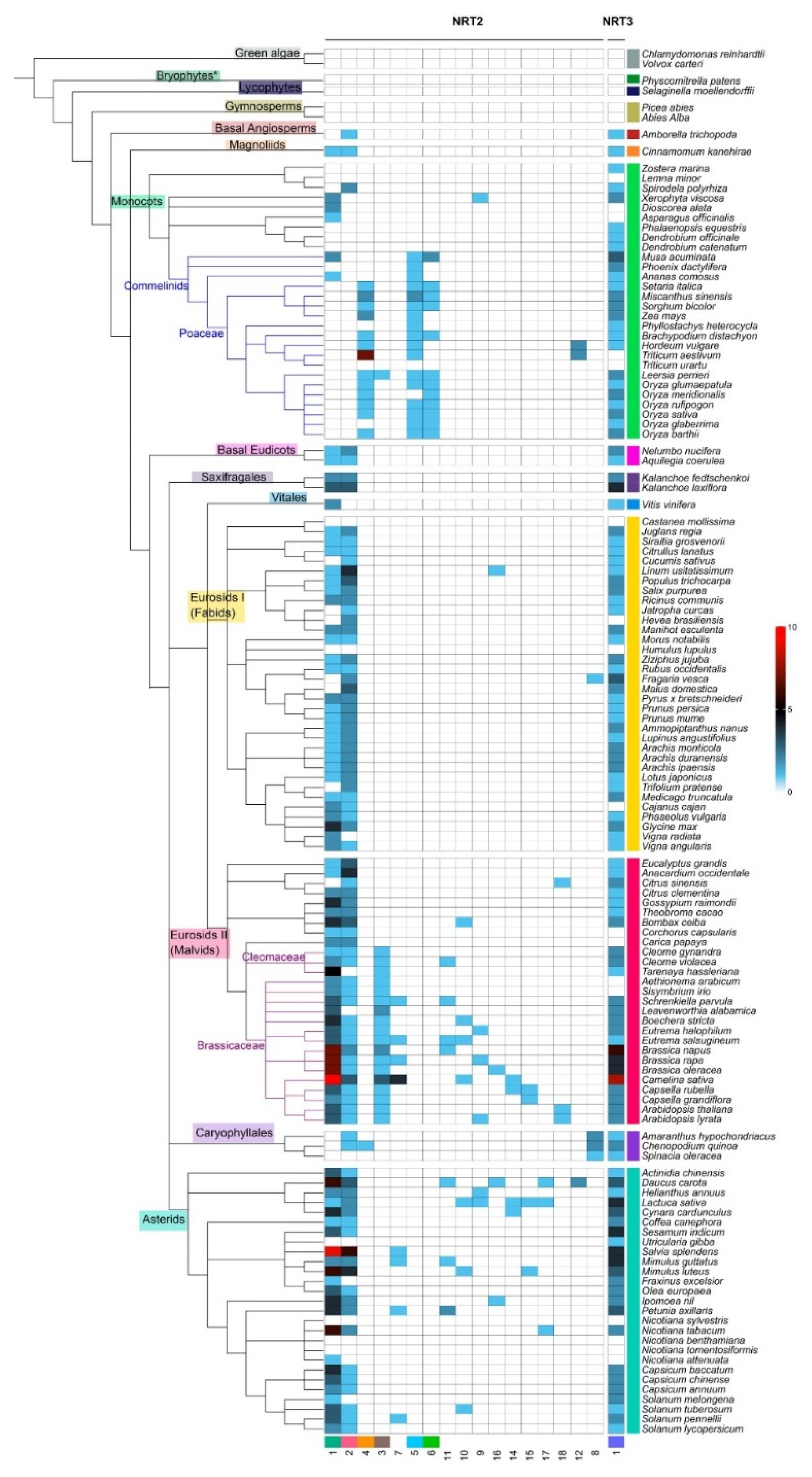
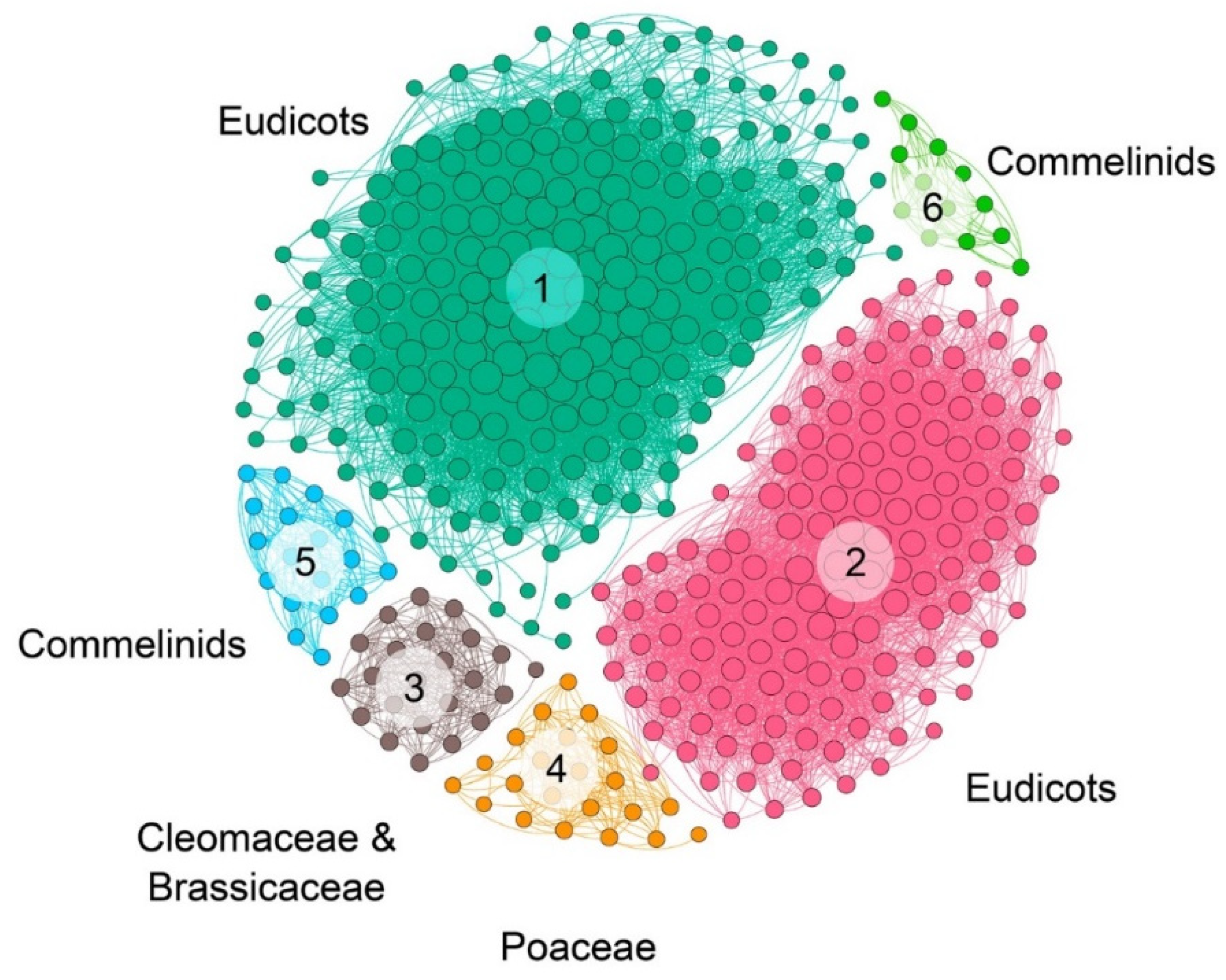
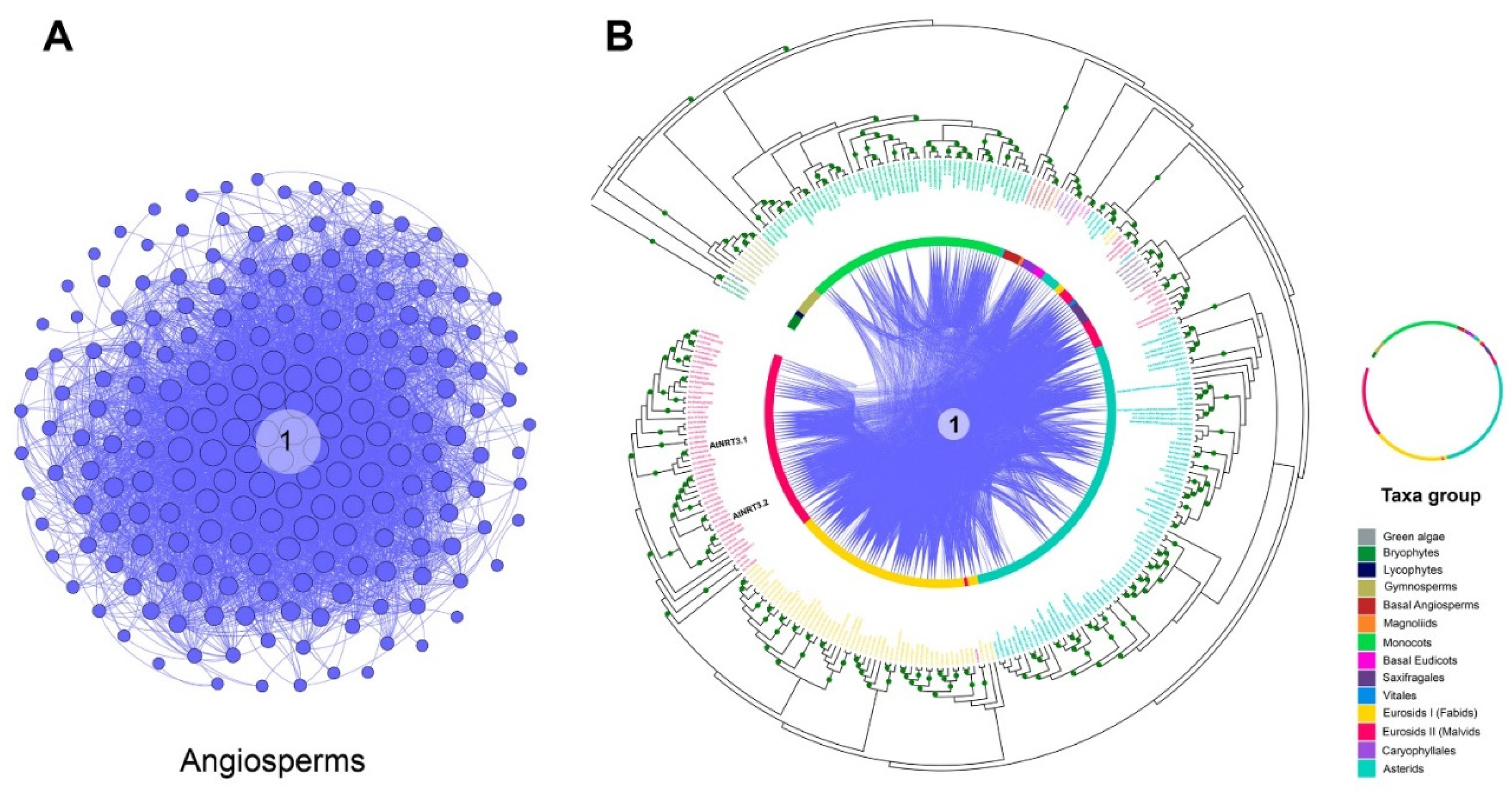
2.2. Microsynteny Analysis of Nitrate Transports
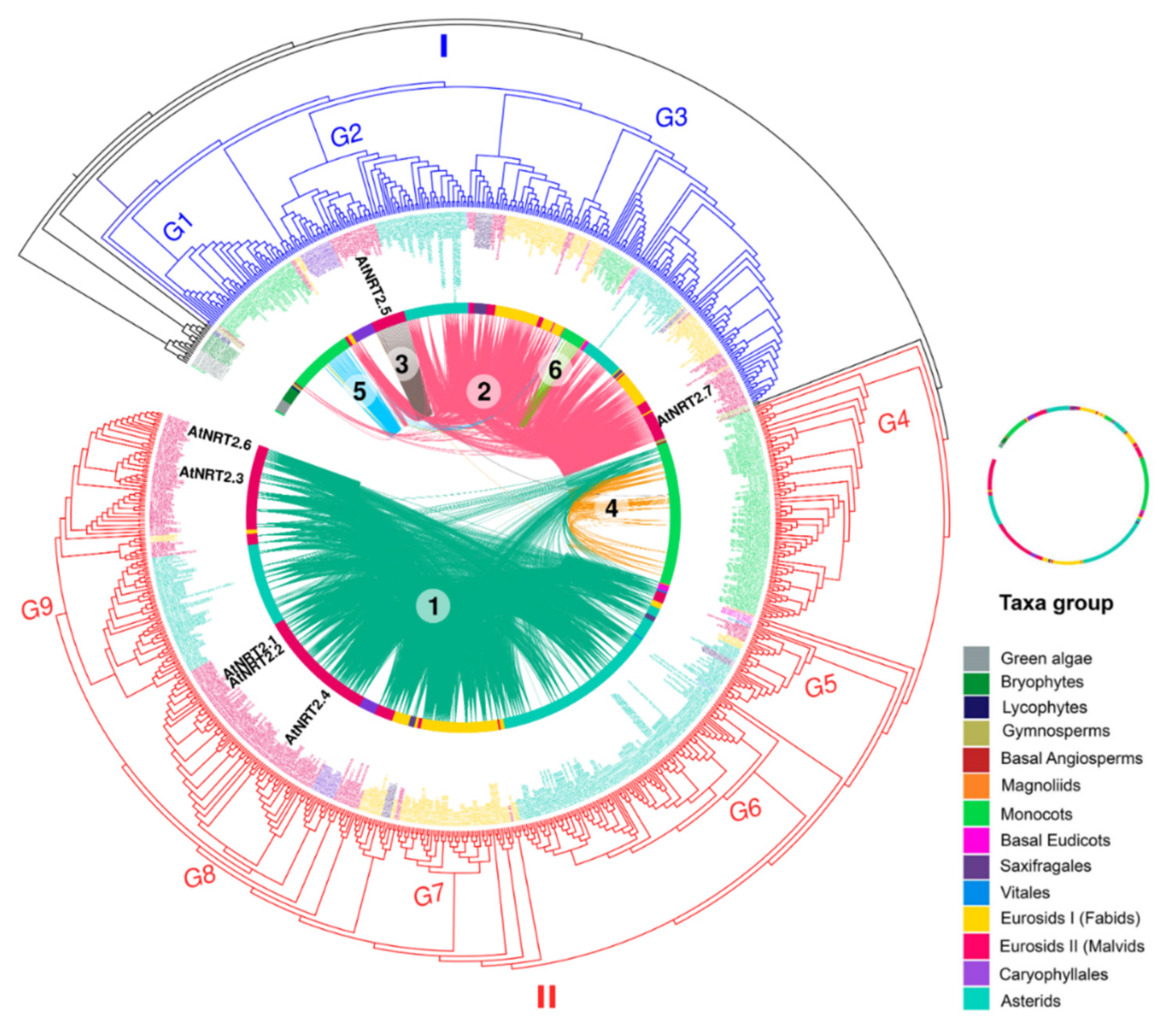
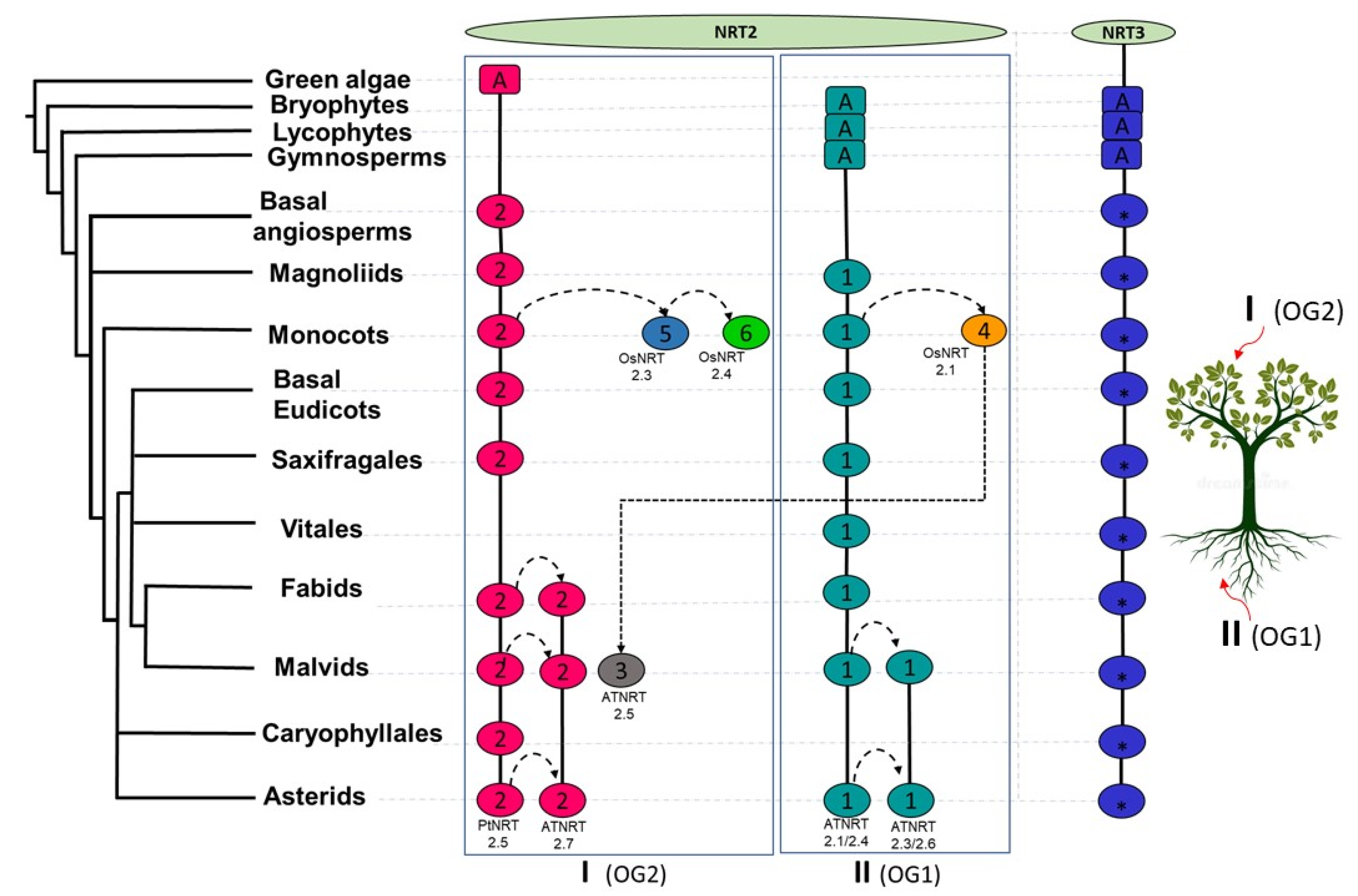
2.3. Orthology Analysis of NRT2 and NRT3
3. Discussion
3.1. Variation in the Number of NRT2 and NRT3 Sequences as a Potential Driver of Habitat Diversity with the Availability of Different N Sources in Vascular Plants
3.2. Variability Analysis into Monocots
3.3. Variability Analysis into Eudicots
3.4. Phylogenomic and Microsynteny Analysis of NRT2 and NRT3 Gene Families in Angiosperms
4. Materials and Methods
4.1. Database Construction
4.2. Phylogenetic Inference
4.3. Synteny Analysis
4.4. Orthology Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vidal, E.A.; Alvarez, J.M.; Araus, V.; Riveras, E.; Brooks, M.D.; Krouk, G.; Ruffel, S.; Lejay, L.; Crawford, N.M.; Coruzzi, G.M.; et al. Nitrate in 2020: Thirty Years from Transport to Signaling Networks. Plant Cell 2020, 32, 2094–2119. [Google Scholar] [CrossRef]
- Fredes, I.; Moreno, S.; Diaz, F.P.; Gutierrez, R.A. Nitrate signaling and the control of Arabidopsis growth and development. Curr. Opin. Plant Biol. 2019, 47, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.; O’Brien, J.A.; Gutiérrez, R. Nitrate and hormonal signaling crosstalk for plant growth and development. Curr. Opin. Plant Biol. 2019, 52, 155–163. [Google Scholar] [CrossRef]
- Dechorgnat, J.; Nguyen, C.T.; Armengaud, P.; Jossier, M.; Diatloff, E.; Filleur, S.; Daniel-Vedele, F. From the soil to the seeds: The long journey of nitrate in plants. J. Exp. Bot. 2010, 62, 1349–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristova, D.; Carré, C.; Pervent, M.; Medici, A.; Kim, G.J.; Scalia, D.; Ruffel, S.; Birnbaum, K.D.; Lacombe, B.; Busch, W.; et al. Combinatorial interaction network of transcriptomic and phenotypic responses to nitrogen and hormones in the Arabidopsis thaliana root. Sci. Signal. 2016, 9, rs13. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N.M.; Glass, A.D. Molecular and physiological aspects of nitrate uptake in plants. Trends Plant Sci. 1998, 3, 389–395. [Google Scholar] [CrossRef]
- Tong, J.F.; Walk, T.C.; Han, P.P.; Chen, L.Y.; Shen, X.J.; Li, Y.S.; Gu, C.M.; Xie, L.H.; Hu, X.J.; Liao, X.; et al. Genome-wide identification and analysis of high-affinity nitrate transporter 2 (NRT2) family genes in rapeseed (Brassica napus L.) and their responses to various stresses. BMC Plant Biol. 2020, 20, 464. [Google Scholar] [CrossRef]
- Tong, Y.; Zhou, J.-J.; Li, Z.; Miller, A.J. A two-component high-affinity nitrate uptake system in barley. Plant J. 2004, 41, 442–450. [Google Scholar] [CrossRef]
- Hua, Y.-P.; Zhou, T.; Song, H.-X.; Guan, C.-Y.; Zhang, Z.-H. Integrated genomic and transcriptomic insights into the two-component high-affinity nitrate transporters in allotetraploid rapeseed. Plant Soil 2018, 427, 245–268. [Google Scholar] [CrossRef]
- Krapp, A.; David, L.C.; Chardin, C.; Girin, T.; Marmagne, A.; Leprince, A.-S.; Chaillou, S.; Ferrario-Méry, S.; Meyer, C.; Daniel-Vedele, F. Nitrate transport and signalling in Arabidopsis. J. Exp. Bot. 2014, 65, 789–798. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Cheng, Y.-H.; Chen, K.-E.; Tsay, Y.-F. Nitrate Transport, Signaling, and Use Efficiency. Annu. Rev. Plant Biol. 2018, 69, 85–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, X.; Xu, C.; Wang, Q. Identification and characterization of the NPF, NRT2 and NRT3 in spinach. Plant Physiol. Biochem. 2020, 158, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zheng, Y.; Feng, H.; Qu, H.; Fan, X.; Yamaji, N.; Ma, J.F.; Xu, G. OsNRT2.4 encodes a dual-affinity nitrate transporter and functions in nitrate-regulated root growth and nitrate distribution in rice. J. Exp. Bot. 2018, 69, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.T.; Guerra, E.; Fernández, E.; Galván, A. Nitrite Reductase Mutants as an Approach to Understanding Nitrate Assimilation in Chlamydomonas reinhardtii. Plant Physiol. 2000, 122, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, B.G. Nitrate transporters in plants: Structure, function and regulation. Biochim. Biophys. Acta (BBA) Biomembr. 2000, 1465, 219–235. [Google Scholar] [CrossRef]
- Niño-González, M.; Novo-Uzal, E.; Richardson, D.N.; Barros, P.M.; Duque, P. More Transporters, More Substrates: The Arabidopsis Major Facilitator Superfamily Revisited. Mol. Plant 2019, 12, 1182–1202. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.S.; Prashant, R.; Kadoo, N.Y.; Upadhyay, A.; Gupta, V.S. Global study of MFS superfamily transporters in Arabidopsis and grapes reveals their functional diversity in plants. Plant Gene 2019, 18, 100179. [Google Scholar] [CrossRef]
- Chopin, F.; Orsel, M.; Dorbe, M.-F.; Chardon, F.; Truong, H.-N.; Miller, A.J.; Krapp, A.; Daniel-Vedele, F. The Arabidopsis ATNRT2.7 Nitrate Transporter Controls Nitrate Content in Seeds. Plant Cell 2007, 19, 1590–1602. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.R.; Tang, Z.; Tan, Y.W.; Zhang, Y.; Luo, B.B.; Yang, M.; Lian, X.M.; Shen, Q.R.; Miller, A.J.; Xu, G.H. Overexpression of a pH-sensitive nitrate transporter in rice increases crop yields. Proc. Natl. Acad. Sci. USA 2016, 113, 7118–7123. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Zhao, J.; Feng, S.; Qiao, K.; Gong, S.; Wang, J.; Zhou, A. Heterologous Expression of Nitrate Assimilation Related-Protein DsNAR2.1/NRT3.1 Affects Uptake of Nitrate and Ammonium in Nitrogen-Starved Arabidopsis. Int. J. Mol. Sci. 2020, 21, 4027. [Google Scholar] [CrossRef]
- Lizama-Gasca, M.G.; Estrada-Tapia, G.; Escalante-Magana, C.A.; Martinez-Estevez, M.; Zepeda-Jazo, I.; Medina-Lara, F.; Echevarria-Machado, I. Cloning and Molecular Characterization of CcNRT2.1/CcNAR2, a Putative Inducible High Affinity Nitrate Transport System in Capsicum chinense Jacq. Roots. Trop. Plant Biol. 2020, 13, 73–90. [Google Scholar] [CrossRef]
- Okamoto, M.; Kumar, A.; Li, W.; Wang, Y.; Siddiqi, M.Y.; Crawford, N.M.; Glass, A.D. High-Affinity Nitrate Transport in Roots of Arabidopsis Depends on Expression of the NAR2-Like Gene AtNRT3.1. Plant Physiol. 2006, 140, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotur, Z.; Mackenzie, N.; Ramesh, S.; Tyerman, S.D.; Kaiser, B.N.; Glass, A.D.M. Nitrate transport capacity of the Arabidopsis thaliana NRT2 family members and their interactions with AtNAR2.1. New Phytol. 2012, 194, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.R.; Naz, M.; Fan, X.R.; Xuan, W.; Miller, A.J.; Xu, G.H. Plant nitrate transporters: From gene function to application. J. Exp. Bot. 2017, 68, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jia, T.; Cui, B.; Song, J. The expression patterns and putative function of nitrate transporter 2.5 in plants. Plant Signal. Behav. 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Lezhneva, L.; Kiba, T.; Feria-Bourrellier, A.B.; Lafouge, F.; Boutet-Mercey, S.; Zoufan, P.; Sakakibara, H.; Daniel-Vedele, F.; Krapp, A. The Arabidopsis nitrate transporter NRT2.5 plays a role in nitrate acquisition and remobilization in nitrogen-starved plants. Plant J. 2014, 80, 230–241. [Google Scholar] [CrossRef]
- Kechid, M.; Desbrosses, G.; Rokhsi, W.; Varoquaux, F.; Djekoun, A.; Touraine, B. The NRT2.5 and NRT2.6 genes are involved in growth promotion of Arabidopsis by the plant growth-promoting rhizobacterium (PGPR) strain Phyllobacterium brassicacearum STM196. New Phytol. 2013, 198, 514–524. [Google Scholar] [CrossRef]
- Reyes-Herrera, P.H.; Muñoz-Baena, L.; Velásquez-Zapata, V.; Patiño, L.; Delgado-Paz, O.A.; Díaz-Diez, C.A.; Navas-Arboleda, A.A.; Cortés, A.J. Inheritance of Rootstock Effects in Avocado (Persea americana Mill.) cv. Hass. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef]
- López-Hernández, F.; Cortés, A.J. Last-Generation Genome–Environment Associations Reveal the Genetic Basis of Heat Tolerance in Common Bean (Phaseolus vulgaris L.). Front. Genet. 2019, 10, 954. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Escudero, M.; Osorio, A.N.; Cortés, A.J. Integrative Pre-Breeding for Biotic Resistance in Forest Trees. Plants 2021, 10, 2022. [Google Scholar] [CrossRef]
- Feng, H.; Fan, X.; Yan, M.; Liu, X.; Miller, A.J.; Xu, G. Multiple roles of nitrate transport accessory protein NAR2 in plants. Plant Signal. Behav. 2011, 6, 1286–1289. [Google Scholar] [CrossRef]
- Bai, H.; Euring, D.; Volmer, K.; Janz, D.; Polle, A. The Nitrate Transporter (NRT) Gene Family in Poplar. PLoS ONE 2013, 8, e72126. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yan, M.; Hu, B.; Priyadarshani, S.V.G.N.; Hou, Z.; Ojolo, S.P.; Xiong, J.; Zhao, H.; Qin, Y. Characterization and the Expression Analysis of Nitrate Transporter (NRT) Gene Family in Pineapple. Trop. Plant Biol. 2018, 11, 177–191. [Google Scholar] [CrossRef]
- Dechorgnat, J.; Francis, K.L.; Dhugga, K.S.; Rafalski, J.A.; Tyerman, S.D.; Kaiser, B.N. Tissue and nitrogen-linked expression profiles of ammonium and nitrate transporters in maize. BMC Plant Biol. 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Li, Y.; Wang, S.; Li, D.; Lv, C.; Xu, R. Characterization of the Nitrate Transporter gene family and functional identification of HvNRT2.1 in barley (Hordeum vulgare L.). PLoS ONE 2020, 15, e0232056. [Google Scholar] [CrossRef]
- Pellizzaro, A.; Planchet, E.; Limami, A.M.; Alibert, B.; Paven, M.-C.M.-L. Nitrate transporters: An overview in legumes. Planta 2017, 246, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Rubio, L.; Díaz-García, J.; Amorim-Silva, V.; Macho, A.P.; Botella, M.A.; Fernández, J.A. Molecular Characterization of ZosmaNRT2, the Putative Sodium Dependent High-Affinity Nitrate Transporter of Zostera marina L. Int. J. Mol. Sci. 2019, 20, 3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, P.L.; Liu, Q.; Li, G.J.; Di, D.W.; Xia, G.M.; Kronzucker, H.J.; Fang, S.; Chu, J.F.; Shi, W.M. TaANR1-TaBG1 and TaWabi5-TaNRT2s/NARs Link ABA Metabolism and Nitrate Acquisition in Wheat Roots. Plant Physiol. 2020, 182, 1440–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, M.M.; Wang, H.; Ahmad, B.; Liu, Y.; Fan, S.; Li, K.; Lei, C.; Shah, K.; Li, S.H.; Zhang, D. Identification and characterization of NRT gene family reveals their critical response to nitrate regulation during adventitious root formation and development in apple rootstock. Sci. Hortic. 2021, 275, 109642. [Google Scholar] [CrossRef]
- von Wittgenstein, N.J.; Le, C.H.; Hawkins, B.J.; Ehlting, J. Evolutionary classification of ammonium, nitrate, and peptide transporters in land plants. BMC Evol. Biol. 2014, 14, 11. [Google Scholar] [CrossRef] [Green Version]
- Li, M.J.; Tian, H.; Gao, Y.J. A genome-wide analysis of NPF and NRT2 transporter gene families in bread wheat provides new insights into the distribution, function, regulation and evolution of nitrate transporters. Plant Soil 2021, 465, 47–63. [Google Scholar] [CrossRef]
- Kelleher, C.T.; Wilkin, J.; Zhuang, J.; Cortés, A.J.; Quintero, A.L.P.; Gallagher, T.F.; Bohlmann, J.; Douglas, C.J.; Ellis, B.E.; Ritland, K. SNP discovery, gene diversity, and linkage disequilibrium in wild populations of Populus tremuloides. Tree Genet. Genomes 2012, 8, 821–829. [Google Scholar] [CrossRef]
- Zhao, T.; Schranz, M.E. Network approaches for plant phylogenomic synteny analysis. Curr. Opin. Plant Biol. 2017, 36, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Restrepo-Montoya, D.; McClean, P.E.; Osorno, J.M. Orthology and synteny analysis of receptor-like kinases “RLK” and receptor-like proteins “RLP” in legumes. BMC Genom. 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Pereira-Santana, A.; Gamboa-Tuz, S.D.; Zhao, T.; Schranz, M.E.; Vinuesa, P.; Bayona, A.; Rodríguez-Zapata, L.C.; Castano, E. Fibrillarin evolution through the Tree of Life: Comparative genomics and microsynteny network analyses provide new insights into the evolutionary history of Fibrillarin. PLoS Comput. Biol. 2020, 16, e1008318. [Google Scholar] [CrossRef]
- Zhao, T.; Schranz, M.E. Network-based microsynteny analysis identifies major differences and genomic outliers in mammalian and angiosperm genomes. Proc. Natl. Acad. Sci. USA 2019, 116, 2165–2174. [Google Scholar] [CrossRef] [Green Version]
- Gamboa-Tuz, S.D.; Pereira-Santana, A.; Zamora-Briseno, J.A.; Castano, E.; Espadas-Gil, F.; Ayala-Sumuano, J.T.; Keb-Llanes, M.A.; Sanchez-Teyer, F.; Rodriguez-Zapata, L.C. Transcriptomics and co-expression networks reveal tissue-specific responses and regulatory hubs under mild and severe drought in papaya (Carica papaya L.). Sci. Rep. 2018, 8, 1–16. [Google Scholar]
- Liu, D.; Hunt, M.; Tsai, I.J. Inferring synteny between genome assemblies: A systematic evaluation. BMC Bioinform. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Gao, B.; Wang, L.; Oliver, M.; Chen, M.; Zhang, J. Evolution of Auxin Response Factors in plants characterized by phylogenomic synteny network analyses. bioRxiv 2019, arXiv:10.1101/603175. [Google Scholar]
- Lee, S.; Choi, S.; Jeon, D.; Kang, Y.; Kim, C. Evolutionary impact of whole genome duplication in Poaceae family. J. Crop Sci. Biotechnol. 2020, 23, 413–425. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Luque, E.; Chamizo-Ampudia, A.; Llamas, A.; Galvan, A.; Fernandez, E. Understanding nitrate assimilation and its regulation in microalgae. Front. Plant Sci. 2015, 6, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, E.; Galvan, A. Inorganic nitrogen assimilation in Chlamydomonas. J. Exp. Bot. 2007, 58, 2279–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, V.A.; Barbazuk, W.B.; Depamphilis, C.W.; Der, J.P.; Leebens-Mack, J.; Ma, H.; Palmer, J.D.; Rounsley, S.; Sankoff, D.; Schuster, S.C.; et al. The Amborella Genome and the Evolution of Flowering Plants. Science 2013, 342, 1241089. [Google Scholar] [CrossRef]
- Wu, S.; Han, B.; Jiao, Y. Genetic Contribution of Paleopolyploidy to Adaptive Evolution in Angiosperms. Mol. Plant 2020, 13, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.-H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ding, G.; Yang, N.; White, P.J.; Ye, X.; Cai, H.; Lu, J.; Shi, L.; Xu, F. Comparative genome and transcriptome analysis unravels key factors of nitrogen use efficiency in Brassica napus L. Plant Cell Environ. 2019, 43, 712–731. [Google Scholar] [CrossRef]
- Olsen, J.L.; Rouzé, P.; Verhelst, B.; Lin, Y.-C.; Bayer, T.; Collen, J.; Dattolo, E.; De Paoli, E.; Dittami, S.; Maumus, F.; et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 2016, 530, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Gruber, N. The Marine Nitrogen Cycle: Overview and Challenges. In Nitrogen in the Marine Environment, 2nd ed.; Capone, D.G., Bronk, D.A., Mulholland, M.R., Carpenter, E.J., Eds.; Academic Press: San Diego, CA, USA, 2008. [Google Scholar]
- Silvera, K.; Santiago, L.; Cushman, J.C.; Winter, K. Crassulacean Acid Metabolism and Epiphytism Linked to Adaptive Radiations in the Orchidaceae. Plant Physiol. 2009, 149, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Liu, X.; Vanneste, K.; Proost, S.; Tsai, W.C.; Liu, K.W.; Chen, L.J.; He, Y.; Xu, Q.; Bian, C.; et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015, 47, 65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.Q.; Xu, Q.; Bian, C.; Tsai, W.C.; Yeh, C.M.; Liu, K.W.; Yoshida, K.; Zhang, L.S.; Chang, S.B.; Chen, F.; et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Wang, X.; Liu, H.; Tian, Y.; Lian, J.; Yang, R.; Hao, S.; Wang, X.; Yang, S.; Li, Q.; et al. The Genome of Dendrobium officinale Illuminates the Biology of the Important Traditional Chinese Orchid Herb. Mol. Plant 2014, 8, 922–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facelli, E.; Smith, S.E.; Smith, F.A. Mycorrhizal symbiosis—Overview and new insights into roles of arbuscular mycorrhizas in agro- and natural ecosystems. Australas. Plant Pathol. 2009, 38, 338–344. [Google Scholar] [CrossRef]
- Hynson, N.A.; Madsen, T.P.; Selosse, M.-A.; Adam, I.K.U.; Ogura-Tsujita, Y.; Roy, M.; Gebauer, G. The Physiological Ecology of Mycoheterotrophy. In Mycoheterotrophy: The Biology of Plants Living on Fungi; Merckx, V., Ed.; Springer: New York, NY, USA, 2013; pp. 297–342. [Google Scholar]
- Koegel, S.; Brule, D.; Wiemken, A.; Boller, T.; Courty, P.E. The effect of different nitrogen sources on the symbiotic interaction between Sorghum bicolor and Glomus intraradices: Expression of plant and fungal genes involved in nitrogen assimilation. Soil Biol. Biochem. 2015, 86, 159–163. [Google Scholar] [CrossRef]
- Müller, A.; George, E.; Gabriel-Neumann, E. The symbiotic recapture of nitrogen from dead mycorrhizal and non-mycorrhizal roots of tomato plants. Plant Soil 2012, 364, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Dearnaley, J.D.W.; Cameron, D. Nitrogen transport in the orchid mycorrhizal symbiosis—Further evidence for a mutualistic association. New Phytol. 2016, 213, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Fochi, V.; Chitarra, W.; Kohler, A.; Voyron, S.; Singan, V.; Lindquist, E.A.; Barry, K.; Girlanda, M.; Grigoriev, I.; Martin, F.; et al. Fungal and plant gene expression in the Tulasnella calospora—Serapias vomeracea symbiosis provides clues about nitrogen pathways in orchid mycorrhizas. New Phytol. 2016, 213, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Li, Q.; Gu, H. Effect of nitrogen deposition and management practices on fine root decomposition in Moso bamboo plantations. Plant Soil 2016, 410, 207–215. [Google Scholar] [CrossRef]
- Zou, N.; Huang, L.; Chen, H.; Huang, X.; Song, Q.; Yang, Q.; Wang, T. Nitrogen form plays an important role in the growth of moso bamboo (Phyllostachys edulis) seedlings. PeerJ 2020, 8, e9938. [Google Scholar] [CrossRef]
- Kerstens, M.H.L.; Schranz, M.E.; Bouwmeester, K. Phylogenomic analysis of the APETALA2 transcription factor sub-family across angiosperms reveals both deep conservation and lineage-specific patterns. Plant J. 2020, 103, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, T.; Luebert, F.; Xiang, Y.; Huang, C.-H.; Hu, Y.; Rees, M.; Frohlich, M.W.; Qi, J.; Weigend, M.; et al. Asterid Phylogenomics/Phylotranscriptomics Uncover Morphological Evolutionary Histories and Support Phylogenetic Placement for Numerous Whole-Genome Duplications. Mol. Biol. Evol. 2020, 37, 3188–3210. [Google Scholar] [CrossRef]
- Carretero-Paulet, L.; Chang, T.H.; Librado, P.; Ibarra-Laclette, E.; Herrera-Estrella, L.; Rozas, J.; Albert, V.A. Genome-Wide Analysis of Adaptive Molecular Evolution in the Carnivorous Plant Utricularia gibba. Genome Biol. Evol. 2015, 7, 444–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leebens-Mack, J.H.; Barker, M.S.; Carpenter, E.J.; Deyholos, M.K.; Gitzendanner, M.A.; Graham, S.W.; Grosse, I.; Li, Z.; Melkonian, M.; Mirarab, S.; et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679. [Google Scholar]
- Wooliver, R.; Pfennigwerth, A.; Bailey, J.K.; Schweitzer, J.A. Plant functional constraints guide macroevolutionary trade-offs in competitive and conservative growth responses to nitrogen. Funct. Ecol. 2016, 30, 1099–1108. [Google Scholar] [CrossRef]
- Ordoñez, J.C.; van Bodegom, P.M.; Witte, J.-P.M.; Wright, I.J.; Reich, P.B.; Aerts, R. A global study of relationships between leaf traits, climate and soil measures of nutrient fertility. Glob. Ecol. Biogeogr. 2009, 18, 137–149. [Google Scholar] [CrossRef]
- Britto, D.T.; Kronzucker, H.J. Ecological significance and complexity of N-source preference in plants. Ann. Bot. 2013, 112, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Kronzucker, H.J.; Siddiqi, M.Y.; Glass, A.D.M. Conifer root discrimination against soil nitrate and the ecology of forest succession. Nature 1997, 385, 59–61. [Google Scholar] [CrossRef]
- Tsujimoto, R.; Yamazaki, H.; Maeda, S.-I.; Omata, T. Distinct Roles of Nitrate and Nitrite in Regulation of Expression of the Nitrate Transport Genes in the Moss Physcomitrella patens. Plant Cell Physiol. 2007, 48, 484–497. [Google Scholar] [CrossRef] [Green Version]
- De Montaigu, A.; Sanz-Luque, E.; Macias, M.I.; Galvan, A.; Fernandez, E. Transcriptional regulation of CDP1 and CYG56 is required for proper NH4+ sensing in Chlamydomonas. J. Exp. Bot. 2010, 62, 1425–1437. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, D.; Okamoto, M.; Vidmar, J.J.; Glass, A.D.M. Regulation of a putative high-affinity nitrate transporter (Nrt2;1At) in roots of Arabidopsis thaliana. Plant J. 1999, 17, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Galvan, A. Nitrate assimilation in Chlamydomonas. Eukaryot Cell 2008, 7, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, S.; Suzaki, T.; Weber, A.P.; Archibald, J.M.; Nozaki, H. Eukaryote-to-eukaryote gene transfer gives rise to genome mosaicism in euglenids. BMC Evol. Biol. 2011, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisecaver, J.H.; Borowsky, A.T.; Tzin, V.; Jander, G.; Kliebenstein, D.J.; Rokas, A. A Global Coexpression Network Approach for Connecting Genes to Specialized Metabolic Pathways in Plants. Plant Cell 2017, 29, 944–959. [Google Scholar] [CrossRef] [Green Version]
- Wisecaver, J.H.; Rokas, A. Fungal metabolic gene clusters-caravans traveling across genomes and environments. Front. Microbiol. 2015, 6, 161. [Google Scholar] [CrossRef] [Green Version]
- Osbourn, A.; Papadopoulou, K.K.; Qi, X.; Field, B.; Wegel, E. Finding and Analyzing Plant Metabolic Gene Clusters. Method Enzymol. 2012, 517, 113–138. [Google Scholar] [CrossRef]
- Nützmann, H.-W.; Osbourn, A. Regulation of metabolic gene clusters in Arabidopsis thaliana. New Phytol. 2014, 205, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Van Hoeck, A.; Horemans, N.; Monsieurs, P.; Cao, H.X.; Vandenhove, H.; Blust, R. The first draft genome of the aquatic model plant Lemna minor opens the route for future stress physiology research and biotechnological applications. Biotechnol. Biofuels 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dluhošová, J.; Ištvánek, J.; Nedělník, J.; Repkova, J. Red Clover (Trifolium pratense) and Zigzag Clover (T. medium)—A Picture of Genomic Similarities and Differences. Front. Plant Sci. 2018, 9, 724. [Google Scholar] [CrossRef]
- Wang, M.H.; Yu, Y.; Haberer, G.; Marri, P.R.; Fan, C.Z.; Goicoechea, J.L.; Zuccolo, A.; Song, X.; Kudrna, D.; Ammiraju, J.S.S.; et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 2014, 46, 982. [Google Scholar] [CrossRef] [Green Version]
- Kotur, Z.; Glass, A.D.M. A 150kDa plasma membrane complex of AtNRT2.5 and AtNAR2.1 is the major contributor to constitutive high-affinity nitrate influx in Arabidopsis thaliana. Plant Cell Environ. 2015, 38, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Plett, D.; Toubia, J.; Garnett, T.; Tester, M.; Kaiser, B.N.; Baumann, U. Dichotomy in the NRT Gene Families of Dicots and Grass Species. PLoS ONE 2010, 5, e15289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.B.; Xu, M.; Zhao, L.M.; Xie, P.; Chen, Y.; Harwood, W.; Xu, G.H.; Fan, X.R.; Miller, A.J. Overexpression of the High-Affinity Nitrate Transporter OsNRT2.3b Driven by Different Promoters in Barley Improves Yield and Nutrient Uptake Balance. Int. J. Mol. Sci. 2020, 21, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabermanesh, K.; Holtham, L.R.; George, J.; Roessner, U.; Boughton, B.; Heuer, S.; Tester, M.; Plett, D.C.; Garnett, T. Transition from a maternal to external nitrogen source in maize seedlings. J. Integr. Plant Biol. 2017, 59, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Ju, L.L.; Deng, G.B.; Liang, J.J.; Zhang, H.L.; Li, Q.; Pan, Z.F.; Yu, M.Q.; Long, H. Structural organization and functional divergence of high isoelectric point α-amylase genes in bread wheat (Triticum aestivum L.) and barley (Hordeum vulgare L.). BMC Genet. 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Feng, S.; Wang, L.; Chen, C.; Lin, Z.; Cheng, H.; Wei, K.; Zhang, C. Cloning and Expressing Analysis of a Nitrogen Transporter 2.5 Gene from Tea Plant Camellia sinensis (L.). J. Tea Sci. 2014, 34, 364–370. [Google Scholar]
- Guo, T.; Xuan, H.; Yang, Y.; Wang, L.; Wei, L.; Wang, Y.; Kang, G. Transcription Analysis of Genes Encoding the Wheat Root Transporter NRT1 and NRT2 Families During Nitrogen Starvation. J. Plant Growth Regul. 2014, 33, 837–848. [Google Scholar] [CrossRef]
- Kiba, T.; Feria-Bourrellier, A.-B.; Lafouge, F.; Lezhneva, L.; Boutet-Mercey, S.; Orsel, M.; Bréhaut, V.; Miller, A.; Dan-iel-Vedele, F.; Sakakibara, H.; et al. The Arabidopsis Nitrate Transporter NRT2.4 Plays a Double Role in Roots and Shoots of Nitrogen-Starved Plants. Plant Cell 2012, 24, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.G.; Zhang, Y.; Tan, Y.W.; Zhang, M.; Zhu, L.L.; Xu, G.H.; Fan, X.R. Agronomic nitrogen-use efficiency of rice can be increased by driving OsNRT2.1 expression with the OsNAR2.1 promoter. Plant Biotechnol. J. 2016, 14, 1705–1715. [Google Scholar] [CrossRef] [Green Version]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern Strategies to Assess and Breed Forest Tree Adaptation to Changing Climate. Front. Plant Sci. 2020, 11, 583323. [Google Scholar] [CrossRef]
- Karunarathne, S.D.; Han, Y.; Zhang, X.Q.; Zhou, G.F.; Hill, C.B.; Chen, K.F.; Angessa, T.; Li, C.D. Genome-Wide Association Study and Identification of Candidate Genes for Nitrogen Use Efficiency in Barley (Hordeum vulgare L.). Front. Plant Sci. 2020, 11, 1361. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zhang, J.; Zhao, Y.; She, M.; Ma, W. Genetic regulation of the traits contributing to wheat nitrogen use efficiency. Plant Sci. 2020, 303, 110759. [Google Scholar] [CrossRef] [PubMed]
- Lupini, A.; Mercati, F.; Araniti, F.; Miller, A.J.; Sunseri, F.; Abenavoli, M.R. NAR2.1/NRT2.1 functional interaction with NO3− and H+ fluxes in high-affinity nitrate transport in maize root regions. Plant Physiol. Biochem. 2016, 102, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.J.; Skeen, P.; Blair, M.W.; Chacon-Sanchez, M.I. Does the Genomic Landscape of Species Divergence in Phaseolus Beans Coerce Parallel Signatures of Adaptation and Domestication? Front. Plant Sci. 2018, 9, 1861. [Google Scholar] [CrossRef] [Green Version]
- Cortés, A.; López-Hernández, F. Harnessing Crop Wild Diversity for Climate Change Adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]
- Cortes, A.J.; Monserrate, F.A.; Ramirez-Villegas, J.; Madrinan, S.; Blair, M.W. Drought Tolerance in Wild Plant Populations: The Case of Common Beans (Phaseolus vulgaris L.). PLoS ONE 2013, 8, 5. [Google Scholar]
- Gong, Y.; Ling, H.; Chen, Y.; Cao, J.; Guo, Z.; Lv, G. N:P stoichiometric changes via species turnover in arid versus saline desert environments. Ecol. Evol. 2020, 10, 6636–6645. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Data from: Ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Zhao, T.; Holmer, R.; de Bruijn, S.; Angenent, G.C.; Burg, H.A.V.D.; Schranz, M.E. Phylogenomic Synteny Network Analysis of MADS-Box Transcription Factor Genes Reveals Lineage-Specific Transpositions, Ancient Tandem Duplications, and Deep Positional Conservation. Plant Cell 2017, 29, 1278–1292. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2011, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosvall, M.; Axelsson, D.; Bergstrom, C.T. The map equation. Eur. Phys. J. 2009, 178, 13–23. [Google Scholar] [CrossRef]
- Rosvall, M.; Bergstrom, C.T. Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. USA 2008, 105, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal 2005, 1695, 1–9. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Available online: https://www.researchgate.net/publication/221297890_Gephi_An_Open_Source_Software_for_Exploring_and_Manipulating_Networks?channel=doi&linkId=0c96053b41bc4075a2000000&showFulltext=true (accessed on 14 October 2021).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Derelle, R.; Philippe, H.; Colbourne, J.K. Broccoli: Combining Phylogenetic and Network Analyses for Orthology Assignment. Mol. Biol. Evol. 2020, 37, 3389–3396. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoghbi-Rodríguez, N.M.; Gamboa-Tuz, S.D.; Pereira-Santana, A.; Rodríguez-Zapata, L.C.; Sánchez-Teyer, L.F.; Echevarría-Machado, I. Phylogenomic and Microsynteny Analysis Provides Evidence of Genome Arrangements of High-Affinity Nitrate Transporter Gene Families of Plants. Int. J. Mol. Sci. 2021, 22, 13036. https://doi.org/10.3390/ijms222313036
Zoghbi-Rodríguez NM, Gamboa-Tuz SD, Pereira-Santana A, Rodríguez-Zapata LC, Sánchez-Teyer LF, Echevarría-Machado I. Phylogenomic and Microsynteny Analysis Provides Evidence of Genome Arrangements of High-Affinity Nitrate Transporter Gene Families of Plants. International Journal of Molecular Sciences. 2021; 22(23):13036. https://doi.org/10.3390/ijms222313036
Chicago/Turabian StyleZoghbi-Rodríguez, Normig M., Samuel David Gamboa-Tuz, Alejandro Pereira-Santana, Luis C. Rodríguez-Zapata, Lorenzo Felipe Sánchez-Teyer, and Ileana Echevarría-Machado. 2021. "Phylogenomic and Microsynteny Analysis Provides Evidence of Genome Arrangements of High-Affinity Nitrate Transporter Gene Families of Plants" International Journal of Molecular Sciences 22, no. 23: 13036. https://doi.org/10.3390/ijms222313036
APA StyleZoghbi-Rodríguez, N. M., Gamboa-Tuz, S. D., Pereira-Santana, A., Rodríguez-Zapata, L. C., Sánchez-Teyer, L. F., & Echevarría-Machado, I. (2021). Phylogenomic and Microsynteny Analysis Provides Evidence of Genome Arrangements of High-Affinity Nitrate Transporter Gene Families of Plants. International Journal of Molecular Sciences, 22(23), 13036. https://doi.org/10.3390/ijms222313036