
Neuroprotective Properties of Quinone Reductase 2 Inhibitor M-11, a 2-Mercaptobenzimidazole Derivative

 , , , , and
, , , , and
Abstract
:1. Introduction
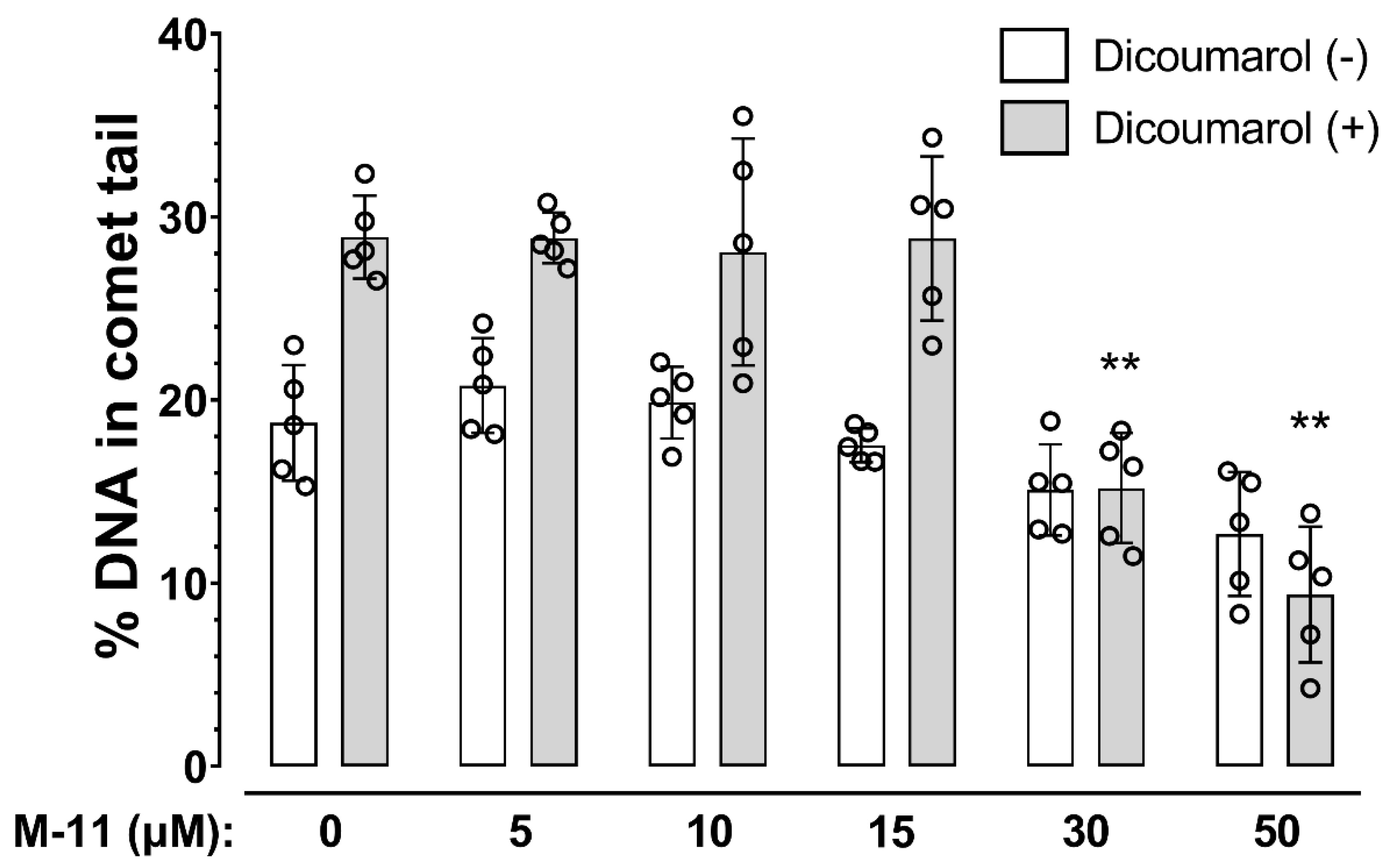
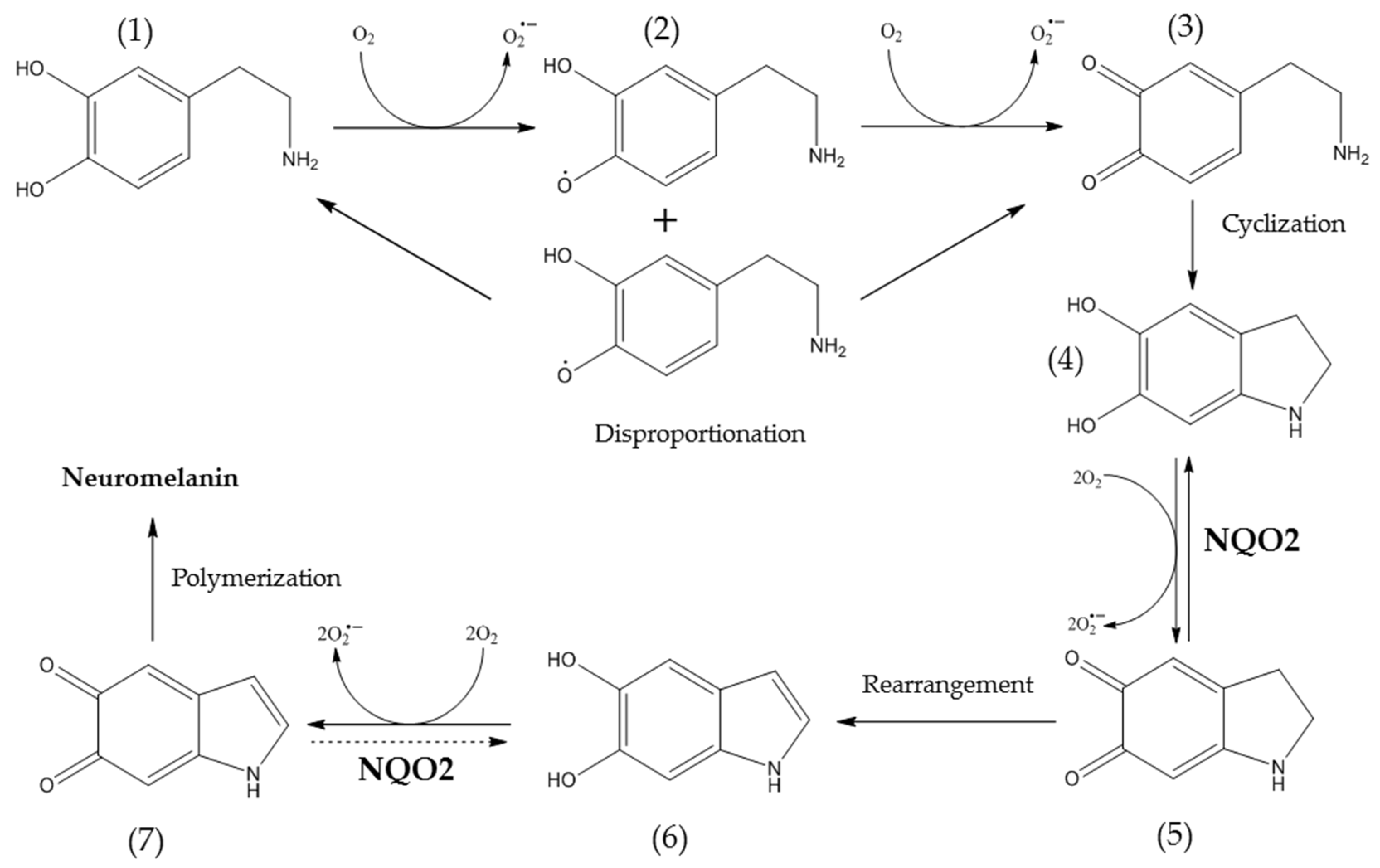
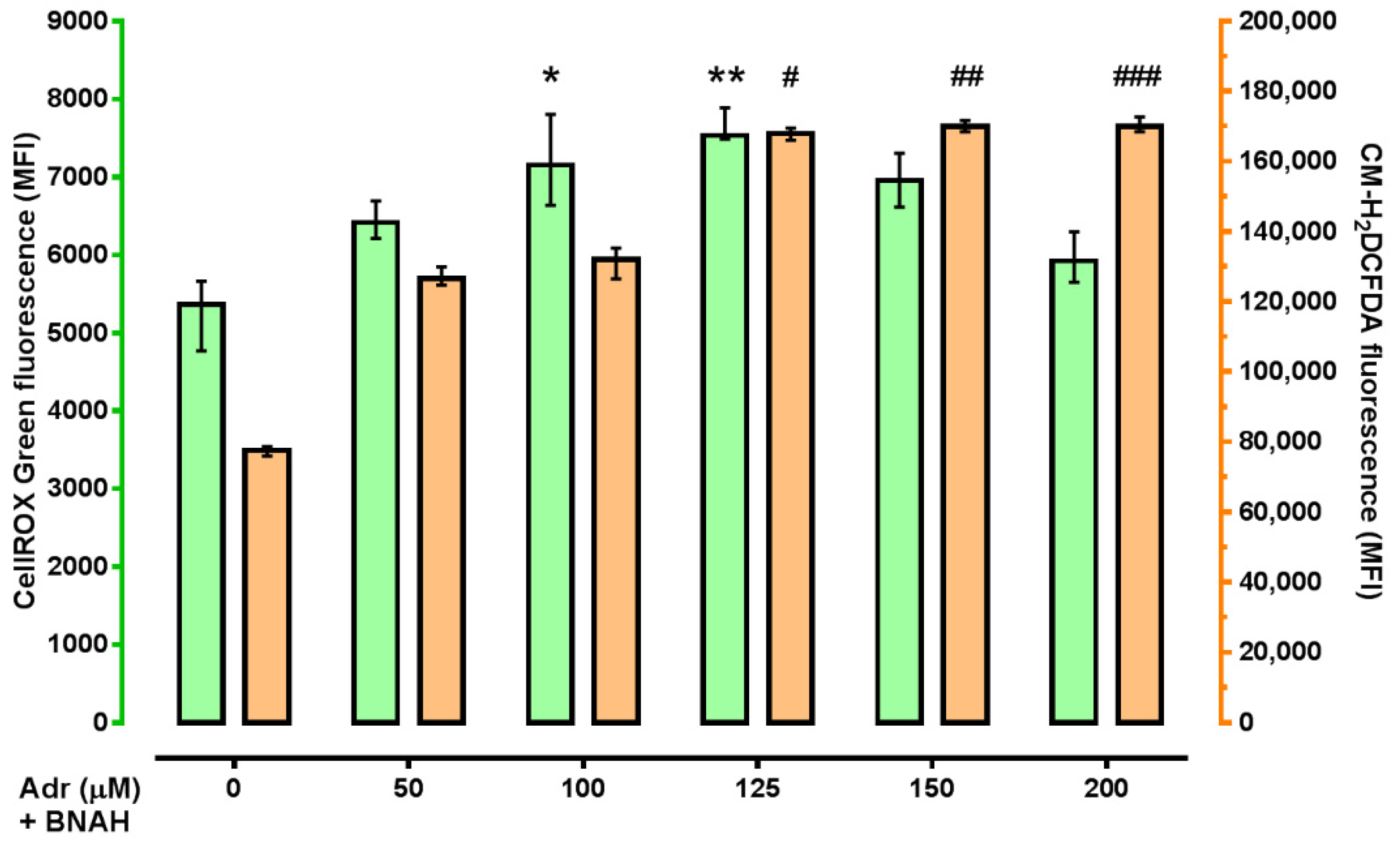
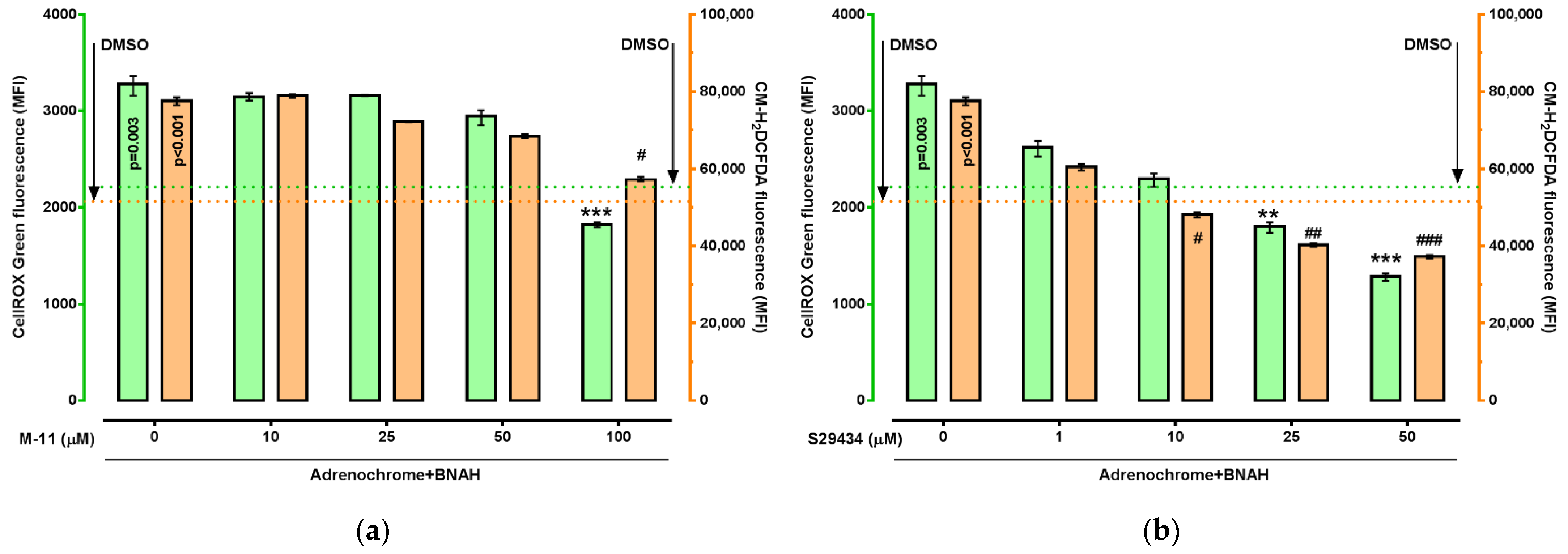
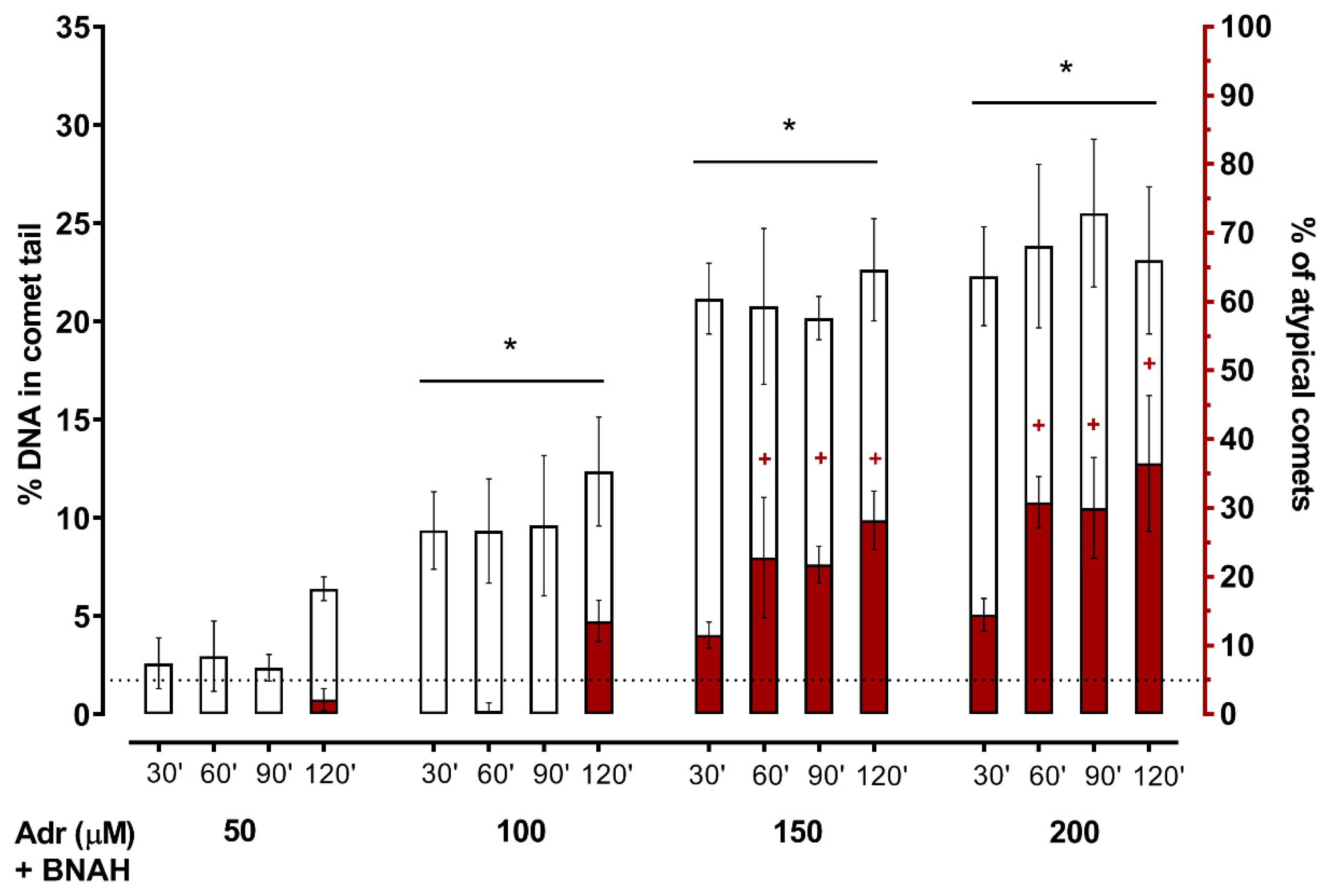
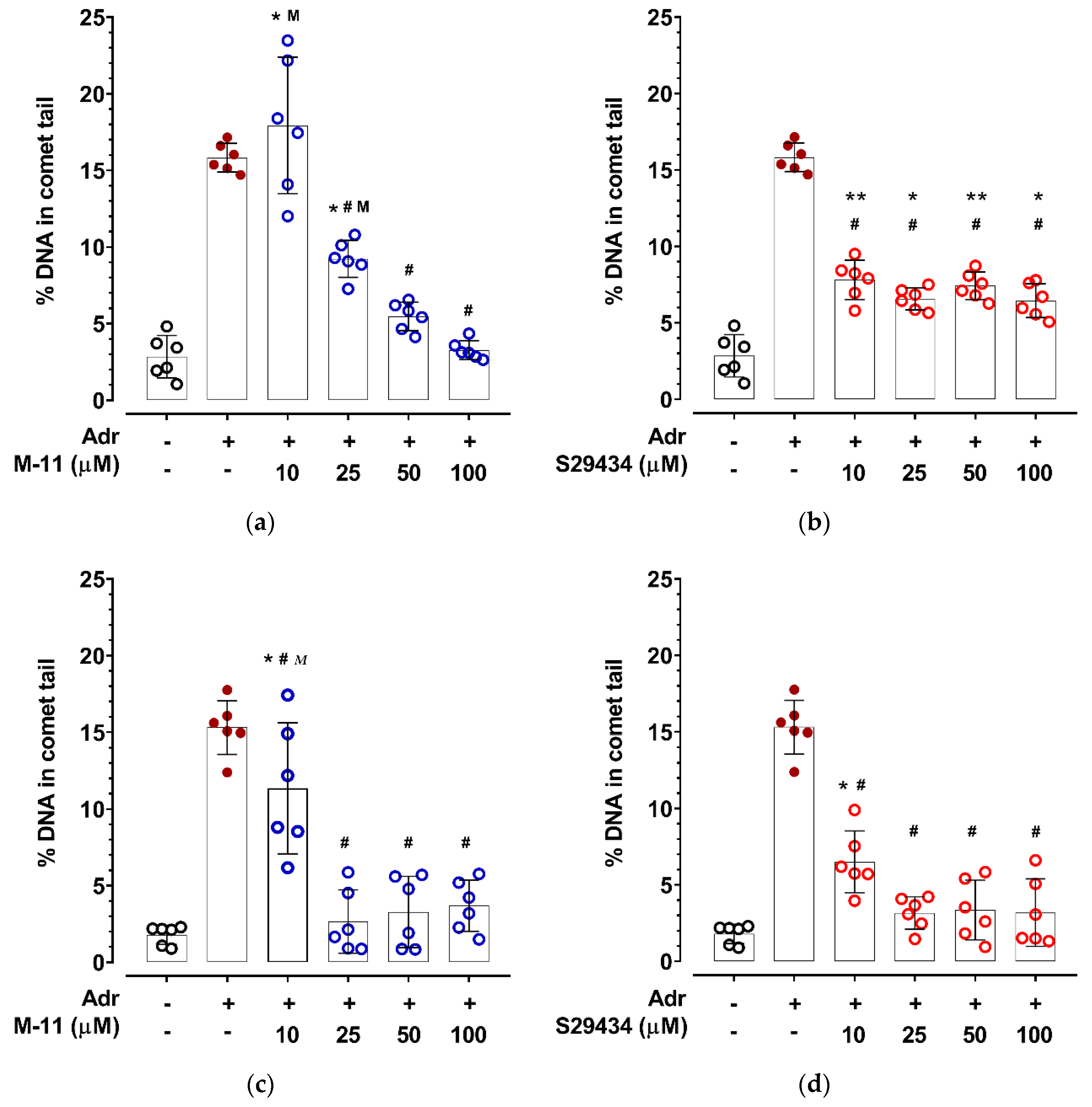
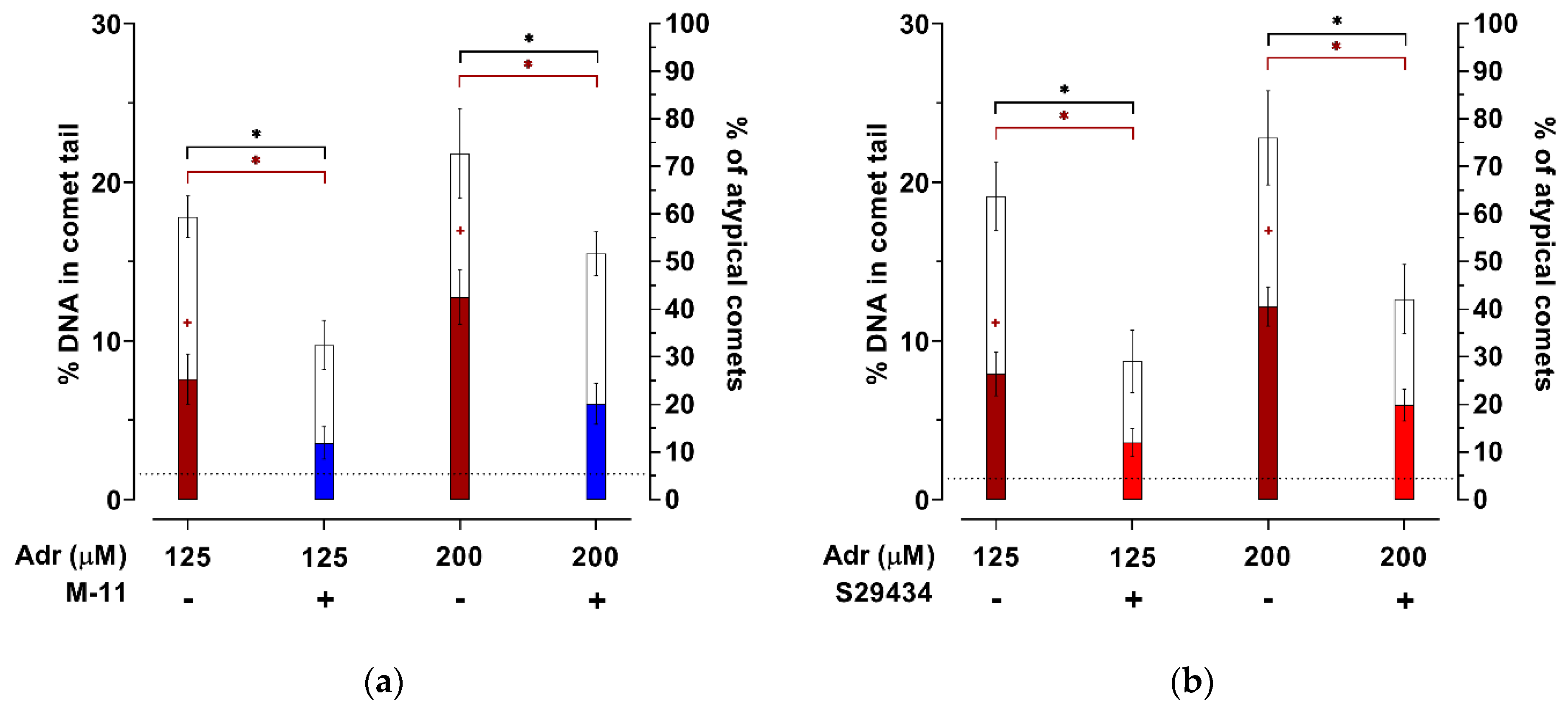
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. HT-22 Immortalized Mouse Hippocampal Neuronal Cell Culture
4.3. Incubation of HT-22 Cells with Substrate, Co-Substrate and Inhibitors of NQO2
4.4. Protein Preparation and Western Blot Analysis
4.5. ROS Measurement
4.6. Apoptosis Assay
4.7. Single-Cell Gel Electrophoresis Assay (Alkaline Comet Assay)
4.8. Experimental Animals
4.9. Ethical Approval
4.10. ICR Mice Bone Marrow Cells
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-Hydroxydopamine: 5-(2-aminoethyl)benzene-1:2:4-triol |
| Afobazole | 5-Ethoxy-2-[2-(morpholino)-ethylthio]benzimidazole dihydrochloride |
| BNAH | 1-Benzyl-1,4-dihydronicotinamide |
| BSA | Bovine serum albumin |
| C3H cells | Murine embryo fibroblast cells |
| CHO cells | Chinese hamster ovary cells |
| CM-H2DCFDA | acetic 2-[3,6-bis(acetyloxy)-2,7-dichloro-9H-xanthen-9-yl]-4-(chloromethyl)benzoic anhydride |
| DMAT | 4,5,6,7-Tetrabromo-N,N-dimethyl-1H-benzimidazol-2-amine |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| EDTA | Ethylenediaminetetraacetic acid |
| EDTA-Na2 | Ethylenediaminetetraacetic acid disodium salt dihydrate |
| EGTA | Ethylenebis(oxyethylenenitrilo)tetraacetic acid |
| EPR | Electron paramagnetic resonance |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HRP | Horseradish peroxidase |
| HT-22 | Immortalized mouse hippocampal cells |
| ICR | Institute of Cancer Research outbred mice |
| K562 cells | Immortalized human myeloid leukemia cells |
| M-11 | 2-[2-(3-Oxomorpholin-4-il)-ethylthio]-5-ethoxy benz-imidazole hydrochloride |
| MAO-A | Amine oxidase [flavin-containing] A |
| MPP+ | N-Methyl-4-phenylpyridine |
| MT1 receptor | Melatonin receptor type 1 |
| MT3 receptor | Melatonin dependent regulatory site of NQO2; melatonin receptor type 3 |
| NADH | Dihydronicotinamide-adenine dinucleotide |
| NADPH | Dihydronicotinamide-adenine dinucleotide phosphate |
| NMNH | 1-(5-O-phosphono-beta-D-ribofuranosyl)-1,4-dihydropyridine-3-carboxamide |
| NQO1 | NAD(P)H dehydrogenase [quinone] 1; QR1 |
| NQO2 | Ribosyldihydronicotinamide dehydrogenase [quinone]; NRH: quinone reductase 2; QR2 |
| NR+ | N-Ribosylnicotinamide |
| NRH | 1-(beta-D-Ribofuranosyl)-1,4-dihydronicotinamide |
| O2•− | Superoxide anion radical |
| OH• | Hydroxyl radical |
| PAGE | Polyacrylamide gel |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| PMSF | Phenylmethylsulfonyl fluoride |
| ROS | Reactive oxygen species |
| RPMI-1640 | Roswell Park Memorial Institute medium |
| S29434 | N-(2-(2-Methoxy-6H-dipyrido[2,3-a:3′,2′-e]pyrrolizin-11-yl)ethyl)furan-2-carboxamide; NMDPEF |
| SDS | Sodium dodecyl sulfate |
| SH-SY5Y cells | Human bone marrow biopsy-derived neuroblastoma cells |
| Sigma1R | Sigma non-opioid intracellular receptor 1, chaperone Sigma1R |
| TBB | 4,5,6,7-Tetrabromobenzotriazole |
| TBBz | 4,5,6,7-Tetrabromobenzimidazole |
| TBHP | tert-Butyl hydroperoxide |
| TBS-T | Tris buffered saline-Tween 20 |
| VAT2 | Synaptic vesicular amine transporter; VMAT2 |
| V-ATPase | V-type proton ATPase |
Appendix A
References
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef] [Green Version]
- Ruz, C.; Alcantud, J.L.; Vives Montero, F.; Duran, R.; Bandres-Ciga, S. Proteotoxicity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5646. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Salvador, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015; p. 905. [Google Scholar]
- Laher, I. Back Matter. In Systems Biology of Free Radicals and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; p. 4178. [Google Scholar]
- Amato, A.; Terzo, S.; Mule, F. Natural Compounds as Beneficial Antioxidant Agents in Neurodegenerative Disorders: A Focus on Alzheimer’s Disease. Antioxidants 2019, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Ebadi, M. Antioxidants as Potential Therapeutics in Neurodegeneration. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 2191–2273. [Google Scholar] [CrossRef]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef] [PubMed]
- Stower, H. Searching for Alzheimer’s disease therapies. Nat. Med. 2018, 24, 894–897. [Google Scholar] [CrossRef]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, Oxidative Stress and Protein-Quinone Modifications in Parkinson’s and Other Neurodegenerative Diseases. Angew. Chem. 2019, 58, 6512–6527. [Google Scholar] [CrossRef]
- Chen, L.; Ding, Y.; Cagniard, B.; Van Laar, A.D.; Mortimer, A.; Chi, W.; Hastings, T.G.; Kang, U.J.; Zhuang, X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 2008, 28, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Manini, P.; Panzella, L.; Napolitano, A.; d’Ischia, M. Oxidation Chemistry of Norepinephrine: Partitioning of the O-Quinone between Competing Cyclization and Chain Breakdown Pathways and Their Roles in Melanin Formation. Chem. Res. Toxicol. 2007, 20, 1549–1555. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buee, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, C.; Murphy, K.P.; Parent, M.; Levine, M.S. The role of dopamine in Huntington’s disease. Prog. Brain Res. 2014, 211, 235–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Y.; Wang, E.A.; Cepeda, C.; Levine, M.S. Dopamine imbalance in Huntington’s disease: A mechanism for the lack of behavioral flexibility. Front. Neurosci. 2013, 7, 114. [Google Scholar] [CrossRef] [Green Version]
- de Diego-Balaguer, R.; Schramm, C.; Rebeix, I.; Dupoux, E.; Durr, A.; Brice, A.; Charles, P.; Cleret de Langavant, L.; Youssov, K.; Verny, C.; et al. COMT Val158Met Polymorphism Modulates Huntington’s Disease Progression. PLoS ONE 2016, 11, e0161106. [Google Scholar] [CrossRef] [Green Version]
- Guillot, T.S.; Miller, G.W. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol. 2009, 39, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Paris, I. Mechanisms of Dopamine Oxidation and Parkinson’s Disease. In Handbook of Neurotoxicity; Kostrzewa, R.M., Ed.; Springer: New York, NY, USA, 2014; pp. 865–883. [Google Scholar] [CrossRef]
- Umek, N.; Gersak, B.; Vintar, N.; Sostaric, M.; Mavri, J. Dopamine Autoxidation Is Controlled by Acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [Green Version]
- Tossounian, M.A.; Zhang, B.; Gout, I. The Writers, Readers, and Erasers in Redox Regulation of GAPDH. Antioxidants 2020, 9, 1288. [Google Scholar] [CrossRef] [PubMed]
- Bucher, M.L.; Barrett, C.W.; Moon, C.J.; Mortimer, A.D.; Burton, E.A.; Greenamyre, J.T.; Hastings, T.G. Acquired dysregulation of dopamine homeostasis reproduces features of Parkinson’s disease. NPJ Parkinson Dis. 2020, 6, 34. [Google Scholar] [CrossRef]
- Eiden, L.E.; Weihe, E. VMAT2: A dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse. Ann. N. Y. Acad. Sci. 2011, 1216, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosharov, E.V. Regulation of DA Homeostasis and Role of VMAT2 in DA-Induced Neurodegeneration. In Handbook of Neurotoxicity; Kostrzewa, R.M., Ed.; Springer: New York, NY, USA, 2014; pp. 973–993. [Google Scholar] [CrossRef]
- Herrera, A.; Munoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nakao, K.; Tanaka, H.; Kitahori, Y.; Tanaka, Y.; Ojika, M.; Ito, S. The Oxidative Pathway to Dopamine-Protein Conjugates and Their Pro-Oxidant Activities: Implications for the Neurodegeneration of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalieri, E.L.; Li, K.M.; Balu, N.; Saeed, M.; Devanesan, P.; Higginbotham, S.; Zhao, J.; Gross, M.L.; Rogan, E.G. Catechol ortho-quinones: The electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis 2002, 23, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.; Saeed, M.; Yang, L.; Beseler, C.; Rogan, E.; Cavalieri, E.L. Formation of dopamine quinone-DNA adducts and their potential role in the etiology of Parkinson’s disease. IUBMB Life 2011, 63, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Nepveu, F.; Calamini, B.; Ferry, G.; Boutin, J.A. Molecular pharmacology of NRH:quinone oxidoreductase 2: A detoxifying enzyme acting as an undercover toxifying enzyme. Mol. Pharmacol. 2020, 98, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Buryanovskyy, L.; Zhang, Z. Quinone reductase 2 is a catechol quinone reductase. J. Biol. Chem. 2008, 283, 23829–23835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindoli, A.; Scutari, G.; Rigobello, M.P. The role of adrenochrome in stimulating the oxidation of catecholamines. Neurotox. Res. 1999, 1, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Song, Y.; Buettner, G.R. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic. Biol. Med. 2010, 49, 919–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Quinone chemistry and toxicity. Toxicol. Appl. Pharmacol. 1992, 112, 2–16. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Cassagnes, L.E.; Chhour, M.; Perio, P.; Sudor, J.; Gayon, R.; Ferry, G.; Boutin, J.A.; Nepveu, F.; Reybier, K. Oxidative stress and neurodegeneration: The possible contribution of quinone reductase 2. Free Radic. Biol. Med. 2018, 120, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Cassagnes, L.E.; Perio, P.; Ferry, G.; Moulharat, N.; Antoine, M.; Gayon, R.; Boutin, J.A.; Nepveu, F.; Reybier, K. In cellulo monitoring of quinone reductase activity and reactive oxygen species production during the redox cycling of 1,2 and 1,4 quinones. Free Radic. Biol. Med. 2015, 89, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Ruszkiewicz, J.; Albrecht, J. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem. Int. 2015, 88, 66–72. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, G.; Peana, M.; Maes, M.; Dadar, M.; Severin, B. The glutathione system in Parkinson’s disease and its progression. Neurosci. Biobehav. Rev. 2021, 120, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Standish, L.J.; Weiss, N.S.; Padowski, J.M.; Kavanagh, T.J.; White, C.C.; Rosenfeld, M.E. Glutathione as a Biomarker in Parkinson’s Disease: Associations with Aging and Disease Severity. Oxidative Med. Cell. Longev. 2016, 2016, 9409363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, T.P.; Bjorklund, M. NQO2 is a reactive oxygen species generating off-target for acetaminophen. Mol. Pharm. 2014, 11, 4395–4404. [Google Scholar] [CrossRef] [Green Version]
- Brain Tissue Expression of NQO2. Available online: https://www.proteinatlas.org/ENSG00000124588-NQO2/tissue (accessed on 1 December 2021).
- Chen, D.; Li, X.; Liu, X.; Liu, X.; Jiang, X.; Du, J.; Wang, Q.; Liang, Y.; Ma, W. NQO2 inhibition relieves reactive oxygen species effects on mouse oocyte meiotic maturation and embryo development. Biol. Reprod. 2017, 97, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yang, X.L.; Holtzclaw, W.D.; Talalay, P. Unexpected genetic and structural relationships of a long-forgotten flavoenzyme to NAD(P)H:quinone reductase (DT-diaphorase). Proc. Natl. Acad. Sci. USA 1997, 94, 1669–1674. [Google Scholar] [CrossRef] [Green Version]
- Foster, C.E.; Bianchet, M.A.; Talalay, P.; Zhao, Q.; Amzel, L.M. Crystal structure of human quinone reductase type 2, a metalloflavoprotein. Biochemistry 1999, 38, 9881–9886. [Google Scholar] [CrossRef] [PubMed]
- Molinari, E.J.; North, P.C.; Dubocovich, M.L. 2-[125I]iodo-5-methoxycarbonylamino-N-acetyltryptamine: A selective radioligand for the characterization of melatonin ML2 binding sites. Eur. J. Pharmacol. 1996, 301, 159–168. [Google Scholar] [CrossRef]
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J. Biol. Chem. 2000, 275, 31311–31317. [Google Scholar] [CrossRef] [Green Version]
- Boutin, J.A.; Ferry, G. Is There Sufficient Evidence that the Melatonin Binding Site MT3 Is Quinone Reductase 2? J. Pharmacol. Exp. Ther. 2019, 368, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Ferry, G.; Hecht, S.; Berger, S.; Moulharat, N.; Coge, F.; Guillaumet, G.; Leclerc, V.; Yous, S.; Delagrange, P.; Boutin, J.A. Old and new inhibitors of quinone reductase 2. Chem. Biol. Interact. 2010, 186, 103–109. [Google Scholar] [CrossRef]
- Leung, K.K.; Shilton, B.H. Binding of DNA-Intercalating Agents to Oxidized and Reduced Quinone Reductase 2. Biochemistry 2015, 54, 7438–7448. [Google Scholar] [CrossRef] [PubMed]
- Buryanovskyy, L.; Fu, Y.; Boyd, M.; Ma, Y.; Hsieh, T.C.; Wu, J.M.; Zhang, Z. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry 2004, 43, 11417–11426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Knox, R.; Sun, X.Z.; Joseph, P.; Jaiswal, A.K.; Zhang, D.; Deng, P.S.; Chen, S. Catalytic properties of NAD(P)H:quinone oxidoreductase-2 (NQO2), a dihydronicotinamide riboside dependent oxidoreductase. Arch. Biochem. Biophys. 1997, 347, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Pegan, S.D.; Sturdy, M.; Ferry, G.; Delagrange, P.; Boutin, J.A.; Mesecar, A.D. X-ray structural studies of quinone reductase 2 nanomolar range inhibitors. Protein Sci. 2011, 20, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghamdi, B.S. The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res. 2018, 96, 1136–1149. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zaki, M.G.; Chen, Z.; Jakova, E.; Ming, Z.; Cayabyab, F.S. Adenosine Signaling and Clathrin-Mediated Endocytosis of Glutamate AMPA Receptors in Delayed Hypoxic Injury in Rat Hippocampus: Role of Casein Kinase 2. Mol. Neurobiol. 2021, 58, 1932–1951. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Agrawal, M.; Dore, S. Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem. Neurosci. 2013, 4, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.G.; Garrick, J.M.; Roque, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxidat. Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsouri, L.; Vizcaychipi, M.P.; McArthur, S.; Harrison, I.; Suarez-Calvet, M.; Lleo, A.; Lloyd, D.G.; Ma, D.; Sastre, M. Prazosin, an alpha(1)-adrenoceptor antagonist, prevents memory deterioration in the APP23 transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Seredenin, S.B.; Voronina, T.A.; Neznamov, G.G.; Blednov Iu, A.; Badyshtov, B.A.; Viglinskaia, I.V.; Kozlovskaia, M.M.; Kolotilinskaia, N.V.; Iarkova, M.A.; Savel’ev, V.L.; et al. The pharmacogenetic concept of the anxio-selective effect. Vestn. Ross. Akad. Med. Nauk 1998, 11, 3–9. [Google Scholar]
- Seredenin, S.B.; Voronin, M.V. Neuroreceptor mechanisms of the afobazole effect. Eksperimental’naia I Klin. Farmakol. 2009, 72, 3–11. [Google Scholar] [CrossRef]
- Seredenin, S.B.; Viglinskaia, A.O.; Mozhaeva, T.; Kolyvanov, G.B.; Litvin, A.A.; Avdiunina, N.I.; Savel’ev, V.L.; Zgerdev, V.P. Afobazole metabolism in rats. Eksperimental’naia I Klin. Farmakol. 2008, 71, 50–52. [Google Scholar] [CrossRef]
- Seredenin, S.B.; Viglinskaia, A.O.; Kolyvanov, G.B.; Litvin, A.A.; Kravtsova, O.; Zherdev, V.P. Pharmacokinetics of afobazole in rats. Eksperimental’naia I Klin. Farmakol. 2007, 70, 59–64. [Google Scholar] [CrossRef]
- Kadnikov, I.A.; Voronin, M.V.; Seredenin, S.B. Afobazole metabolite M-11 inhibits quinone reductase 2. Farmakokinet. I Farm. 2018, 3, 27–30. [Google Scholar] [CrossRef]
- Kadnikov, I.A.; Voronin, M.V.; Seredenin, S.B. Cytoprotective Effect of Afobazole and Its Main Metabolite M-11. Bull. Exp. Biol. Med. 2015, 159, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Voronin, M.V.; Kadnikov, I.A. Contribution of Sigma-1 receptor to cytoprotective effect of afobazole. Pharmacol. Res. Perspect. 2016, 4, e00273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadnikov, I.A.; Voronkov, D.N.; Voronin, M.V.; Seredenin, S.B. Analysis of Quinone Reductase 2 Implication in Mechanism of Antiparkinsonian Action of Afobazole. Neurochem. J. 2020, 14, 227–234. [Google Scholar] [CrossRef]
- Vasiliou, V.; Ross, D.; Nebert, D.W. Update of the NAD(P)H:quinone oxidoreductase (NQO) gene family. Hum. Genom. 2006, 2, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.R. Measuring oxidative damage to DNA and its repair with the comet assay. Biochim. Et Biophys. Acta 2014, 1840, 794–800. [Google Scholar] [CrossRef]
- Fang, L.; Neutzner, A.; Turtschi, S.; Flammer, J.; Mozaffarieh, M. Comet assay as an indirect measure of systemic oxidative stress. J. Vis. Exp. JoVE 2015, 99, e52763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, X.; Wang, Z.; Klaunig, J.E. Alkaline Comet Assay for Assessing DNA Damage in Individual Cells. Curr Protoc Toxicol. 2015, 65, 3.12.1–3.12.11. [Google Scholar] [CrossRef]
- Lorenzo, Y.; Costa, S.; Collins, A.R.; Azqueta, A. The comet assay, DNA damage, DNA repair and cytotoxicity: Hedgehogs are not always dead. Mutagenesis 2013, 28, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Zhanataev, A.K.; Anisina, E.A.; Chayka, Z.V.; Miroshkina, I.A.; Durnev, A.D. The phenomenon of atypical DNA comets. Cell Tissue Biol. 2017, 11, 286–292. [Google Scholar] [CrossRef]
- Boutin, J.A.; Bouillaud, F.; Janda, E.; Gacsalyi, I.; Guillaumet, G.; Hirsch, E.C.; Kane, D.A.; Nepveu, F.; Reybier, K.; Dupuis, P.; et al. S29434, a Quinone Reductase 2 Inhibitor: Main Biochemical and Cellular Characterization. Mol. Pharmacol. 2019, 95, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Chomarat, P.; Coge, F.; Guenin, S.P.; Mailliet, F.; Vella, F.; Mallet, C.; Giraudet, S.; Nagel, N.; Leonce, S.; Ferry, G.; et al. Cellular knock-down of quinone reductase 2: A laborious road to successful inhibition by RNA interference. Biochimie 2007, 89, 1264–1275. [Google Scholar] [CrossRef]
- Park, S.Y.; Jung, W.J.; Kang, J.S.; Kim, C.M.; Park, G.; Choi, Y.W. Neuroprotective effects of alpha-iso-cubebene against glutamate-induced damage in the HT22 hippocampal neuronal cell line. Int. J. Mol. Med. 2015, 35, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, C.R.; Hu, Q.H.; Bresnahan, C.G.; Hati, S.; Bhattacharyya, S. Cyclic Changes in Active Site Polarization and Dynamics Drive the ‘Ping-pong’ Kinetics in NRH:Quinone Oxidoreductase 2: An Insight from QM/MM Simulations. ACS Catal. 2018, 8, 12015–12029. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Buryanovskyy, L.; Zhang, Z. Crystal structure of quinone reductase 2 in complex with cancer prodrug CB1954. Biochem. Biophys. Res. Commun. 2005, 336, 332–338. [Google Scholar] [CrossRef]
- Foster, C.E.; Bianchet, M.A.; Talalay, P.; Faig, M.; Amzel, L.M. Structures of mammalian cytosolic quinone reductases. Free Radic. Biol. Med. 2000, 29, 241–245. [Google Scholar] [CrossRef]
- Reybier, K.; Perio, P.; Ferry, G.; Bouajila, J.; Delagrange, P.; Boutin, J.A.; Nepveu, F. Insights into the redox cycle of human quinone reductase 2. Free Radic. Res. 2011, 45, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Soriano, M.E.; Arduini, I.; Mammi, S.; Bubacco, L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: Implications for mitochondrial dysfunction in Parkinson disease. Biochim. Et Biophys. Acta 2010, 1802, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, V.M.; Silva, R.; Ferreira, L.M.; Branco, P.S.; Carvalho, F.; Bastos, M.L.; Carvalho, R.A.; Carvalho, M.; Remiao, F. Oxidation process of adrenaline in freshly isolated rat cardiomyocytes: Formation of adrenochrome, quinoproteins, and GSH adduct. Chem. Res. Toxicol. 2007, 20, 1183–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayat, F.; Sonavane, M.; Makarov, M.V. The Biochemical Pathways of Nicotinamide-Derived Pyridones. Int. J. Mol. Sci. 2021, 22, 1145. [Google Scholar] [CrossRef]
- Pyle, J.R.; Sy Piecco, K.W.E.; Vicente, J.R.; Chen, J. In Situ Sensing of Reactive Oxygen Species on Dye-Stained Single DNA Molecules under Illumination. Langmuir 2019, 35, 11308–11314. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific Catalog. Available online: https://www.thermofisher.com/order/catalog/product/C6827?SID=srch-srp-C6827 (accessed on 1 December 2021).
- Choi, H.; Yang, Z.; Weisshaar, J.C. Single-cell, real-time detection of oxidative stress induced in Escherichia coli by the antimicrobial peptide CM15. Proc. Natl. Acad. Sci. USA 2015, 112, E303–E310. [Google Scholar] [CrossRef] [Green Version]
- McBee, M.E.; Chionh, Y.H.; Sharaf, M.L.; Ho, P.; Cai, M.W.; Dedon, P.C. Production of Superoxide in Bacteria Is Stress- and Cell State-Dependent: A Gating-Optimized Flow Cytometry Method that Minimizes ROS Measurement Artifacts with Fluorescent Dyes. Front. Microbiol. 2017, 8, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppen, G.; Azqueta, A.; Pourrut, B.; Brunborg, G.; Collins, A.R.; Langie, S.A.S. The next three decades of the comet assay: A report of the 11th International Comet Assay Workshop. Mutagenesis 2017, 32, 397–408. [Google Scholar] [CrossRef]
- Milena, R.; Ninoslav, Đ.; Jevrosima, S.; Marko, A.; Stoimir, K.; Stefan, D.; Zoran, S. The Investigation of DNA Damage Induced by Adrenaline in Human Lymphocytes In Vitro/Ispitivanja Oštećenja DNK Izazvanih Adrenalinom U Limfocitima Čoveka In Vitro. Acta Vet. 2014, 64, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Okamoto, Y.; Aoki, A.; Jinno, H. Catecholamine oxidation-mediated transcriptional inhibition in Mn neurotoxicity. J. Toxicol. Sci. 2020, 45, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.A.; Young, A.J.; Gilmore, I.T.; Morris, A.; Bilton, R.F. Measurement of menadione-mediated DNA damage in human lymphocytes using the comet assay. Free Radic. Res. 1997, 26, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Hulea, S.; Kummerow, F.A. Effects of adrenochrome and epinephrine on human arterial endothelial cells in vitro. Res. Commun. Mol. Pathol. Pharmacol. 1995, 89, 111–126. [Google Scholar]
- Janda, E.; Parafati, M.; Aprigliano, S.; Carresi, C.; Visalli, V.; Sacco, I.; Ventrice, D.; Mega, T.; Vadala, N.; Rinaldi, S.; et al. The antidote effect of quinone oxidoreductase 2 inhibitor against paraquat-induced toxicity in vitro and in vivo. Br. J. Pharmacol. 2013, 168, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppede, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Iskander, K.; Li, J.; Han, S.; Zheng, B.; Jaiswal, A.K. NQO1 and NQO2 regulation of humoral immunity and autoimmunity. J. Biol. Chem. 2006, 281, 30917–30924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, D.J.; Iskander, K.; Gaikwad, A.; Arin, M.; Roop, D.R.; Knox, R.; Barrios, R.; Jaiswal, A.K. Disruption of dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) leads to myeloid hyperplasia of bone marrow and decreased sensitivity to menadione toxicity. J. Biol. Chem. 2002, 277, 46131–46139. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lascala, A.; Carresi, C.; Parafati, M.; Aprigliano, S.; Russo, V.; Savoia, C.; Ziviani, E.; Musolino, V.; Morani, F.; et al. Parkinsonian toxin-induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: Implications for neuroprotection. Autophagy 2015, 11, 1063–1080. [Google Scholar] [CrossRef] [PubMed]
- Benoit, C.E.; Bastianetto, S.; Brouillette, J.; Tse, Y.; Boutin, J.A.; Delagrange, P.; Wong, T.; Sarret, P.; Quirion, R. Loss of quinone reductase 2 function selectively facilitates learning behaviors. J. Neurosci. 2010, 30, 12690–12700. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Nakai, M. Increased hippocampal quinone reductase 2 in Alzheimer’s disease. Neurosci. Lett. 2011, 502, 10–12. [Google Scholar] [CrossRef]
- Wang, W.; Le, W.D.; Pan, T.; Stringer, J.L.; Jaiswal, A.K. Association of NRH:quinone oxidoreductase 2 gene promoter polymorphism with higher gene expression and increased susceptibility to Parkinson’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Mozhaeva, T.Y.; Yarkova, M.A.; Lezina, V.P.; Seredenin, S.B. Synthesis and pharmacological activity of the main metabolite of afobazole and its analogs. Pharm. Chem. J. 2011, 45, 147. [Google Scholar] [CrossRef]
- Wiegant, F.A.C.; Limandjaja, G.; de Poot, S.A.H.; Bayda, L.A.; Vorontsova, O.N.; Zenina, T.A.; Langelaar-Makkinje, M.; Post, J.A.; Wikman, G. Plant adaptogens activate cellular adaptive mechanisms by causing mild damage. In Adaptation Biology and Medicine: Health Potentials; Lukyanova, L., Takeda, N., Singal, P.K., Eds.; Narosa Publishers: New Dehli, India, 2008; Volume 5, pp. 319–332. [Google Scholar]
- GOST 33215-2014. Guidelines for Accommodation and Care of Animals. Environment, Housing and Management. In Interstate Counsil for Standartization, Metrology and Certification; Standartinform: Moscow, Russia, 2016; Available online: https://protect.gost.ru/document.aspx?control=7&id=202494 (accessed on 1 December 2021).
- National Research Council (U.S.); Committee for the Update of the Guide for the Care and Use of Laboratory Animals; Institute for Laboratory Animal Research (U.S.); National Academies Press (U.S.). Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; p. 220. [Google Scholar]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptotic Stages (% of Cells) | ||
|---|---|---|
| Early | Late | |
| 0.3% DMSO | 2.7 ± 0.3 | 0.2 ± 0.1 |
| Adrenochrome/BNAH | 30.9 ± 2.5 *** | 41.4 ± 0.01 *** |
| M-11 (100 μM) + Adrenochrome/BNAH | 32.3 ± 4.8 | 35.7 ± 0.5 ### |
| S29434 (25 μM) + Adrenochrome/BNAH | 32.5 ± 2.2 | 19.4 ± 0.9 ### |
| M-11 (100 μM) | 2.3 ± 0.8 | 0.5 ± 0.07 |
| S29434 (25 μM) | 2.8 ± 0.09 | 0.6 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voronin, M.V.; Kadnikov, I.A.; Zainullina, L.F.; Logvinov, I.O.; Verbovaya, E.R.; Antipova, T.A.; Vakhitova, Y.V.; Seredenin, S.B. Neuroprotective Properties of Quinone Reductase 2 Inhibitor M-11, a 2-Mercaptobenzimidazole Derivative. Int. J. Mol. Sci. 2021, 22, 13061. https://doi.org/10.3390/ijms222313061
Voronin MV, Kadnikov IA, Zainullina LF, Logvinov IO, Verbovaya ER, Antipova TA, Vakhitova YV, Seredenin SB. Neuroprotective Properties of Quinone Reductase 2 Inhibitor M-11, a 2-Mercaptobenzimidazole Derivative. International Journal of Molecular Sciences. 2021; 22(23):13061. https://doi.org/10.3390/ijms222313061
Chicago/Turabian StyleVoronin, Mikhail V., Ilya A. Kadnikov, Liana F. Zainullina, Ilya O. Logvinov, Ekaterina R. Verbovaya, Tatyana A. Antipova, Yulia V. Vakhitova, and Sergei B. Seredenin. 2021. "Neuroprotective Properties of Quinone Reductase 2 Inhibitor M-11, a 2-Mercaptobenzimidazole Derivative" International Journal of Molecular Sciences 22, no. 23: 13061. https://doi.org/10.3390/ijms222313061
APA StyleVoronin, M. V., Kadnikov, I. A., Zainullina, L. F., Logvinov, I. O., Verbovaya, E. R., Antipova, T. A., Vakhitova, Y. V., & Seredenin, S. B. (2021). Neuroprotective Properties of Quinone Reductase 2 Inhibitor M-11, a 2-Mercaptobenzimidazole Derivative. International Journal of Molecular Sciences, 22(23), 13061. https://doi.org/10.3390/ijms222313061