
NLRP3 Inflammasome in Diabetic Cardiomyopathy and Exercise Intervention


Abstract
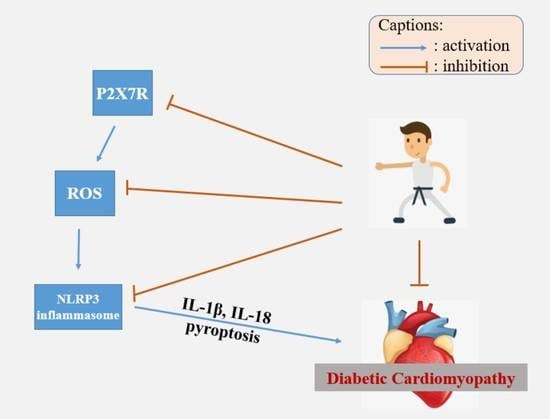
:
1. NLRP3 Inflammasome Biology and Pyroptosis
2. The NLRP3 Inflammasome in the Development of DCM
2.1. DCM, a Severe Complication of Diabetes
2.2. The NLRP3 Inflammasome and Diabetes
2.3. NLRP3 Inflammasome and DCM
2.4. The NLRP3 Inflammasome as the Link between Diabetes, DCM and Heart Failure
2.5. The P2X7 Receptor, NLRP3 Inflammasome and DCM
3. Exercise Intervention for Diabetic Cardiomyopathy
3.1. Exercise Intervention to Regulate the NLRP3 Inflammasome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | Model | Exercise Regimen | Main Findings | Ref |
|---|---|---|---|---|
| Rats | SD, male | Acute treadmill running, 45, 90 or 120 min | Myocardium NLRP3↑, IL-1β↑ following 45 min exercise and during recovery | [64] |
| Human | Healthy, young, male | (1) Acute moderate intensity: 50% HRmax for 30 min + 70% HRmax for 40 min, Nordic walking; (2) Acute high intensity, 70% HRmax for 30 min + 90% HRmax for 40 min, Nordic walking. | (1) No change in PBMCs NLRP3 mRNA, or serum IL-1β and IL-18; (2) PBMCs NLRP3 mRNA↑, serum IL-1β and IL-18↑. | [4] |
| Human | Non-obese, female/male | (1) 2-h exercise, 60% VO2max, cycling (2) 1.5-h exercise, 70% VO2max, cycling | No change in adipose IL-18 mRNA following exercise, or 2 h/10 h during recovery | [66] |
| Mice | C57BL/6, male, HFD | 80% VO2max, treadmill running, 30 min/day, 5 times/week, 10 weeks. | Adipose mRNA of IL-1β↓, TNF-α↓, IL-18↓ | [69] |
| Human | Healthy, young, male | (1) Moderate intensity: 50% HRmax for 30 min + 70% HRmax for 40 min, Nordic walking, 3 days/week, 3 months; (2) High intensity, 70% HRmax for 30 min + 90% HRmax for 40 min, Nordic walking, 3 days/week, 3 months. | (1) PBMCs NLRP3 mRNA↓, serum IL-1β and IL-18↓; (2) PBMCs NLRP3 mRNA↑, serum IL-1β and IL-18↑. | [4] |
| Human | Obese, male and female | High-intensity, 70% VO2max, rowing, 30 min/day, 3 days/week, 8 weeks. | Adipose IL-18 mRNA↓ after training. No change in plasma IL-18 after training. | [66] |
| Human | Obese, male and female | Hypocaloric diet & moderate-intensity, 65–75% HR, aerobic and resistance, 3–5 days/week, 16 weeks. | peripheral blood ASC mRNA↓, MCP-1↓, MIP-1β↓ | [67] |
| Human | Elderly, male and female | Resistance exercise (leg press, biceps curl, pec deck), 60–80% 1RM, 2 sessions/week, 8 weeks. | PBMCs NLRP3↓, caspase-1/pro-caspase-1↓ | [68] |
| Mice | C57BL/6, male, HFD | Isometric strength training, 3 min, 3 series with 1 min break, 5 times/week | Adipose NLRP3↓, serum IL-18↓ | [69] |
3.2. Exercise Intervention to Alleviate DCM
3.3. Exercise Intervention for DCM through the NLRP3 Inflammasome
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; Da, R.F.J.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global Estimates of Diabetes Prevalence for 2017 and Projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hua, Y.; Li, X.; Arslan, I.M.; Zhang, W.; Meng, G. Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 2020, 11, 42. [Google Scholar] [CrossRef]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 8, 519. [Google Scholar] [CrossRef] [Green Version]
- Abkenar, I.K.; Rahmani-Nia, F.; Lombardi, G. The Effects of Acute and Chronic Aerobic Activity on the Signaling Pathway of the Inflammasome NLRP3 Complex in Young Men. Medicina 2019, 55, 105. [Google Scholar] [CrossRef] [Green Version]
- Hordern, M.D.; Coombes, J.S.; Cooney, L.M.; Jeffriess, L.; Prins, J.B.; Marwick, T.H. Effects of exercise intervention on myocardial function in type 2 diabetes. Heart 2009, 95, 1343–1349. [Google Scholar] [CrossRef]
- Kar, S.; Shahshahan, H.R.; Hackfort, B.T.; Yadav, S.K.; Yadav, R.; Kambis, T.N.; Lefer, D.J.; Mishra, P.K. Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxidants 2019, 8, 638. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018, 419, 8–19. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, S.; Sagara, J. Regulatory molecules involved in inflammasome formation with special reference to a key mediator protein, ASC. Semin. Immunopathol. 2007, 29, 231–238. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 Inflammasome: A Sensor for Metabolic Danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.-W.; Zhang, J.; Li, X.; Wang, Y.; Fu, Y.-H.; Gao, X.-Y. A new research hot spot: The role of NLRP3 inflammasome activation, a key step in pyroptosis, in diabetes and diabetic complications. Life Sci. 2020, 240, 117138. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Vigano, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [Green Version]
- Gulsin, G.S.; Athithan, L.; McCann, G.P. Diabetic cardiomyopathy: Prevalence, determinants and potential treatments. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819834869. [Google Scholar] [CrossRef] [PubMed]
- Lundbaek, K. Diabetic angiopathy: A specific vascular disease. Lancet 1954, 266, 377–379. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, D.; Zhang, Y.; Li, M.; Wang, C. Wogonin attenuates diabetic cardiomyopathy through its anti-inflammatory and anti-oxidative properties. Mol. Cell. Endocrinol. 2016, 428, 101–108. [Google Scholar] [CrossRef]
- Zile, M.R.; Brutsaert, D.L. New concepts in diastolic dysfunction and diastolic heart failure: Part I: Diagnosis, prognosis, and measurements of diastolic function. Circulation 2002, 105, 1387–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Kozarez, I.; Adams, V.; Mangner, N.; Höllriegel, R.; Erbs, S.; Linke, A.; Möbius-Winkler, S.; Thiery, J.; Kratzsch, J.; et al. Age-related effects of exercise training on diastolic function in heart failure with reduced ejection fraction: The Leipzig Exercise Intervention in Chronic Heart Failure and Aging (LEICA) Diastolic Dysfunction Study. Eur. Hear. J. 2012, 33, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Hayat, S.A.; Patel, B.; Khattar, R.S.; Malik, R. Diabetic cardiomyopathy: Mechanisms, diagnosis and treatment. Clin. Sci. 2004, 107, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Carlos, D.; Costa, F.R.C.; Pereira, C.A.; Rocha, F.A.; Yaochite, J.N.U.; Oliveira, G.G.; Carneiro, F.S.; Tostes, R.C.; Ramos, S.G.; Zamboni, D.S.; et al. Mitochondrial DNA Activates the NLRP3 Inflammasome and Predisposes to Type 1 Diabetes in Murine Model. Front. Immunol. 2017, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.S.; Bruder-Nascimento, T.; Pereira, C.A.; Zanotto, C.Z.; Prado, D.S.; Silva, J.F.; Rassi, D.M.; Foss-Freitas, M.C.; Alves-Filho, J.C.; Carlos, D.; et al. NLRP3 Inflammasome and Mineralocorticoid Receptors Are Associated with Vascular Dysfunction in Type 2 Diabetes Mellitus. Cells 2019, 8, 1595. [Google Scholar] [CrossRef] [Green Version]
- Jankauskas, S.S.; Kansakar, U.; Varzideh, F.; Wilson, S.; Mone, P.; Lombardi, A.; Gambardella, J.; Santulli, G. Heart failure in diabetes. Metabolism 2021, 125, 154910. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Li, B.; Zhu, X.; Lai, X. Klotho improves diabetic cardiomyopathy by suppressing the NLRP3 inflammasome pathway. Life Sci. 2019, 234, 116773. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, Y.; Zhang, M.; An, F. Rosuvastatin Alleviates Diabetic Cardiomyopathy by Inhibiting NLRP3 Inflammasome and MAPK Pathways in a Type 2 Diabetes Rat Model. Cardiovasc. Drugs Ther. 2013, 28, 33–43. [Google Scholar] [CrossRef]
- Wang, X.; Pan, J.; Liu, H.; Zhang, M.; Liu, D.; Lu, L.; Tian, J.; Liu, M.; Jin, T.; An, F. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019, 221, 249–258. [Google Scholar] [CrossRef]
- Suetomi, T.; Miyamoto, S.; Brown, J.H. Inflammation in nonischemic heart disease: Initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am. J. Physiol. Circ. Physiol. 2019, 317, H877–H890. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the studies of left ventricular dysfunction (SOLVD). J. Am. Coll. Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef]
- Pedersen, B.K. Anti-inflammatory effects of exercise: Role in diabetes and cardiovascular disease. Eur. J. Clin. Investig. 2017, 47, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Wohlford, G.F.; Van Tassell, B.W.; Billingsley, H.E.; Kadariya, D.; Canada, J.M.; Carbone, S.; Mihalick, V.L.; Bonaventura, A.; Vecchie, A.; Chiabrando, J.G.; et al. Phase 1B, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects with NYHA II–III Systolic Heart Failure. J Cardiovasc. Pharmacol. 2020, 77, 49–60. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.A.; Da Silva, J.H.M.; Ferreira, D.N.M.; Fidalgo-Neto, A.A.; Teixeira, P.C.N.; De Souza, C.A.M.; Caffarena, E.R.; De Freitas, M.S. Structural and Molecular Modeling Features of P2X Receptors. Int. J. Mol. Sci. 2014, 15, 4531–4549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [CrossRef]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Dal Susino, M.; Melchiorri, L.; Baricordi, O.R.; Di Virgilio, F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J. Immunol. 1997, 159, 1451–1458. [Google Scholar] [PubMed]
- Franceschini, A.; Capece, M.; Chiozzi, P.; Falzoni, S.; Sanz, J.M.; Sarti, A.C.; Bonora, M.; Pinton, P.; Di Virgilio, F. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 2015, 29, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Solle, M.; Labasi, J.; Perregaux, D.G.; Stam, E.; Petrushova, N.; Koller, B.H.; Griffiths, R.J.; Gabel, C.A. Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem. 2001, 276, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Lenertz, L.Y.; Gavala, M.L.; Hill, L.M.; Bertics, P.J. Cell signaling via the P2X7 nucleotide receptor: Linkage to ROS production, gene transcription, and receptor trafficking. Purinergic Signal. 2009, 5, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontanils, U.; Seil, M.; Pochet, S.; El Ouaaliti, M.; Garcia-Marcos, M.; Dehaye, J.; Marino, A. Stimulation by P2X7 receptors of calcium-dependent production of reactive oxygen species (ROS) in rat submandibular glands. Biochim. Biophys. Acta 2010, 1800, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.; Yerbury, J.; Sluyter, R. P2X7 Receptor Activation Induces Reactive Oxygen Species Formation and Cell Death in Murine EOC13 Microglia. Mediat. Inflamm. 2013, 2013, 1–18. [Google Scholar] [CrossRef]
- Tozzi, M.; Hansen, J.B.; Novak, I. Pannexin-1 mediated ATP release in adipocytes is sensitive to glucose and insulin and modulates lipolysis and macrophage migration. Acta Physiol. 2020, 228, e13360. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Boyce, A.K.; Swayne, L.A. P2X7 receptor cross-talk regulates ATP-induced pannexin 1 internalization. Biochem. J. 2017, 474, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Behura, A.; Kumar, A.; Naik, L.; Swain, A.; Das, M.; Sarangi, S.S.; Dokania, P.; Dirisala, V.R.; Bhutia, S.K.; et al. P2X7 receptor in multifaceted cellular signalling and its relevance as a potential therapeutic target in different diseases. Eur. J. Pharmacol. 2021, 906, 174235. [Google Scholar] [CrossRef]
- Peng, K.; Liu, L.; Wei, D.; Lv, Y.; Wang, G.; Xiong, W.; Wang, X.; Altaf, A.; Wang, L.; Dangheng, W.; et al. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int. J. Mol. Med. 2015, 35, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.; Turner, C.M.; Elliott, J.I.; McDaid, J.; Hewitt, R.; Smith, J.; Pickering, M.; Whitehouse, D.L.; Cook, H.T.; Burnstock, G.; et al. P2X7 Deficiency Attenuates Renal Injury in Experimental Glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 1275–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiansen, S.B.; Skovsted, G.F.; Berchtold, L.A.; Radziwon-Balicka, A.; Dreisig, K.; Edvinsson, L.; Sheykhzade, M.; Haanes, K.A. Role of pannexin and adenosine triphosphate (ATP) following myocardial ischemia/reperfusion. Scand. Cardiovasc. J. 2018, 52, 340–343. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Wang, K.; Liang, X.; Wang, W.; Hu, X.; Huang, Z.; Wang, Y. Aerobic Exercise Ameliorates Myocardial Inflammation, Fibrosis and Apoptosis in High-Fat-Diet Rats by Inhibiting P2X7 Purinergic Receptors. Front. Physiol. 2019, 10, 1286. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Y.; Li, H.; Shen, L.; Chang, Q.; Pan, L.; Hong, S.; Yin, X. H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. J. Cell. Mol. Med. 2018, 22, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Salvadó, L.; Barroso, E.; Vázquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef]
- Wei, M.; Gibbons, L.W.; Kampert, J.B.; Nichaman, M.Z.; Blair, S.N. Low Cardiorespiratory Fitness and Physical Inactivity as Predictors of Mortality in Men with Type 2 Diabetes. Ann. Intern. Med. 2000, 132, 605–611. [Google Scholar] [CrossRef]
- Boule, N.G.; Haddad, E.; Kenny, G.P.; Wells, G.A.; Sigal, R.J. Effects of exercise on glycemic control and body mass in type 2 diabetes mellitus: A meta-analysis of controlled clinical trials. JAMA 2001, 286, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, D.; Ribeiro, P.A.; Kramer, C.K.; Leitao, C.B.; Zucatti, A.T.; Azevedo, M.J.; Gross, J.L.; Ribeiro, J.P.; Schaan, B.D. Physical activity advice only or structured exercise training and association with HbA1c levels in type 2 diabetes: A systematic review and meta-analysis. JAMA 2011, 305, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Miao, W.; Ma, J.; Xv, Z.; Bo, H.; Li, J.; Zhang, Y.; Ji, L.L. Acute Exercise-Induced Mitochondrial Stress Triggers an Inflammatory Response in the Myocardium via NLRP3 Inflammasome Activation with Mitophagy. Oxidative Med. Cell. Longev. 2016, 2016, 19871499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Leick, L.; Lindegaard, B.; Stensvold, D.; Plomgaard, P.; Saltin, B.; Pilegaard, H. Adipose Tissue Interleukin-18 mRNA and Plasma Interleukin-18: Effect of Obesity and Exercise. Obesity 2007, 15, 356–363. [Google Scholar] [CrossRef]
- Barrón-Cabrera, E.; González-Becerra, K.; Rosales-Chávez, G.; Mora-Jiménez, A.; Hernández-Cañaveral, I.; Martínez-López, E. Low-grade chronic inflammation is attenuated by exercise training in obese adults through down-regulation of ASC gene in peripheral blood: A pilot study. Genes Nutr. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Mejías-Peña, Y.; Estébanez, B.; Miguelez, P.R.; Fernandez-Gonzalo, R.; Almar, M.; De Paz, J.A.; González-Gallego, J.; Cuevas, M.J. Impact of resistance training on the autophagy-inflammation-apoptosis crosstalk in elderly subjects. Aging 2017, 9, 408–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardare, C.; Krüger, K.; Liebisch, G.; Seimetz, M.; Couturier, A.; Ringseis, R.; Wilhelm, J.; Weissmann, N.; Eder, K.; Mooren, F.-C. Endurance and Resistance Training Affect High Fat Diet-Induced Increase of Ceramides, Inflammasome Expression, and Systemic Inflammation in Mice. J. Diabetes Res. 2015, 2016, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisafulli, A.; Pagliaro, P.; Roberto, S.; Cugusi, L.; Mercuro, G.; Lazou, A.; Beauloye, C.; Bertrand, L.; Hausenloy, D.J.; Aragno, M.; et al. Diabetic Cardiomyopathy and Ischemic Heart Disease: Prevention and Therapy by Exercise and Conditioning. Int. J. Mol. Sci. 2020, 21, 2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association (ADA). 4. Lifestyle Management: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S38–S50. [Google Scholar] [CrossRef] [Green Version]
- Hansen, F.D.; Coninx, K.; Dendale, F.P. The EAPC EXPERT tool. Eur. Heart J. 2017, 38, 2318–2320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, S.; Cai, Y.; Xie, K.; Zhang, W.; Zheng, F. Effects of combined aerobic and resistance training on the glycolipid metabolism and inflammation levels in type 2 diabetes mellitus. J. Phys. Ther. Sci. 2015, 27, 2365–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.B.; Stampfer, M.J.; Solomon, C.; Liu, S.; Colditz, G.A.; Speizer, F.E.; Willett, W.C.; Manson, J.E. Physical activity and risk for cardiovascular events in diabetic women. Ann. Intern. Med. 2001, 134, 96–105. [Google Scholar] [CrossRef]
- Li, S.; Culver, B.; Ren, J. Benefit and risk of exercise on myocardial function in diabetes. Pharmacol. Res. 2003, 48, 127–132. [Google Scholar] [CrossRef]
- Epp, R.A.; Susser, S.E.; Morissette, M.P.; Kehler, D.S.; Jassal, D.S.; Duhamel, T.A. Exercise training prevents the development of cardiac dysfunction in the low-dose streptozotocin diabetic rats fed a high-fat diet. Can. J. Physiol. Pharmacol. 2013, 91, 80–89. [Google Scholar] [CrossRef]
- Lund, J.; Hafstad, A.D.; Boardman, N.T.; Rossvoll, L.; Rolim, N.P.; Ahmed, M.S.; Florholmen, G.; Attramadal, H.; Wisløff, U.; Larsen, T.S.; et al. Exercise training promotes cardioprotection through oxygen-sparing action in high fat-fed mice. Am. J. Physiol. Circ. Physiol. 2015, 308, H823–H829. [Google Scholar] [CrossRef]
- Schuster, I.; Vinet, A.; Karpoff, L.; Startun, A.; Jourdan, N.; Dauzat, M.; Nottin, S.; Perez-Martin, A. Diastolic Dysfunction and Intraventricular Dyssynchrony Are Restored by Low Intensity Exercise Training in Obese Men. Obesity 2012, 20, 134–140. [Google Scholar] [CrossRef]
- Brassard, P.; Legault, S.; Garneau, C.; Bogaty, P.; Dumesnil, J.-G.; Poirier, P. Normalization of Diastolic Dysfunction in Type 2 Diabetics after Exercise Training. Med. Sci. Sports Exerc. 2007, 39, 1896–1901. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Perrea, D.; Iliadis, F.; Angelopoulou, N.; Liapis, C.; Alevizos, M. Exercise Reduces Resistin and Inflammatory Cytokines in Patients with Type 2 Diabetes. Diabetes Care 2007, 30, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, Y.; LaVoy, E.; Umetani, M.; Hong, J.; Park, Y. Physical activity protects NLRP3 inflammasome-associated coronary vascular dysfunction in obese mice. Physiol. Rep. 2018, 6, e13738. [Google Scholar] [CrossRef]
- Li, Y.; Xu, P.; Wang, Y.; Zhang, J.; Yang, M.; Chang, Y.; Zheng, P.; Huang, H.; Cao, X. Different Intensity Exercise Preconditions Affect Cardiac Function of Exhausted Rats through Regulating TXNIP/TRX/NF-kBp65/NLRP3 Inflammatory Pathways. Evid. Based Complement Altern. Med. 2020, 2020, 5809298. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Ding, S. NLRP3 Inflammasome in Diabetic Cardiomyopathy and Exercise Intervention. Int. J. Mol. Sci. 2021, 22, 13228. https://doi.org/10.3390/ijms222413228
Sun Y, Ding S. NLRP3 Inflammasome in Diabetic Cardiomyopathy and Exercise Intervention. International Journal of Molecular Sciences. 2021; 22(24):13228. https://doi.org/10.3390/ijms222413228
Chicago/Turabian StyleSun, Yi, and Shuzhe Ding. 2021. "NLRP3 Inflammasome in Diabetic Cardiomyopathy and Exercise Intervention" International Journal of Molecular Sciences 22, no. 24: 13228. https://doi.org/10.3390/ijms222413228
APA StyleSun, Y., & Ding, S. (2021). NLRP3 Inflammasome in Diabetic Cardiomyopathy and Exercise Intervention. International Journal of Molecular Sciences, 22(24), 13228. https://doi.org/10.3390/ijms222413228