
Roles of the Na+/H+ Exchanger Isoform 1 and Urokinase in Prostate Cancer Cell Migration and Invasion

 , , , and
, , , and 
Abstract
:1. Introduction
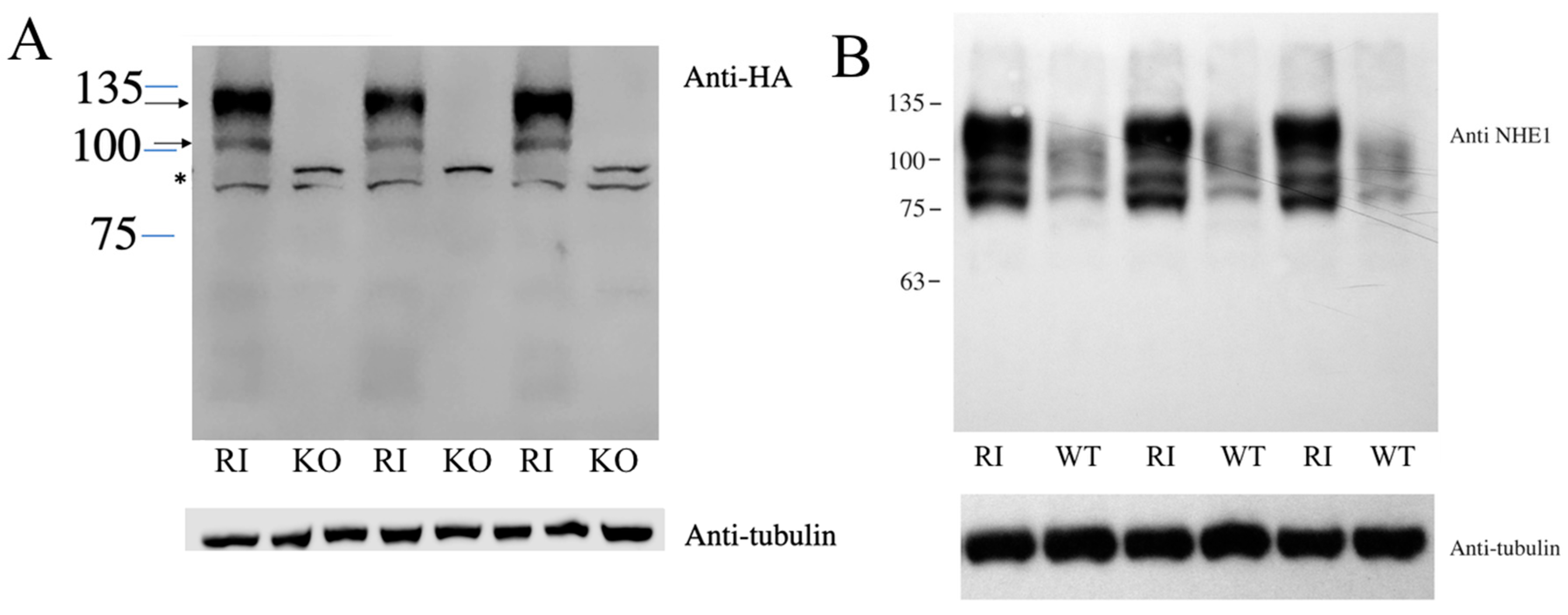
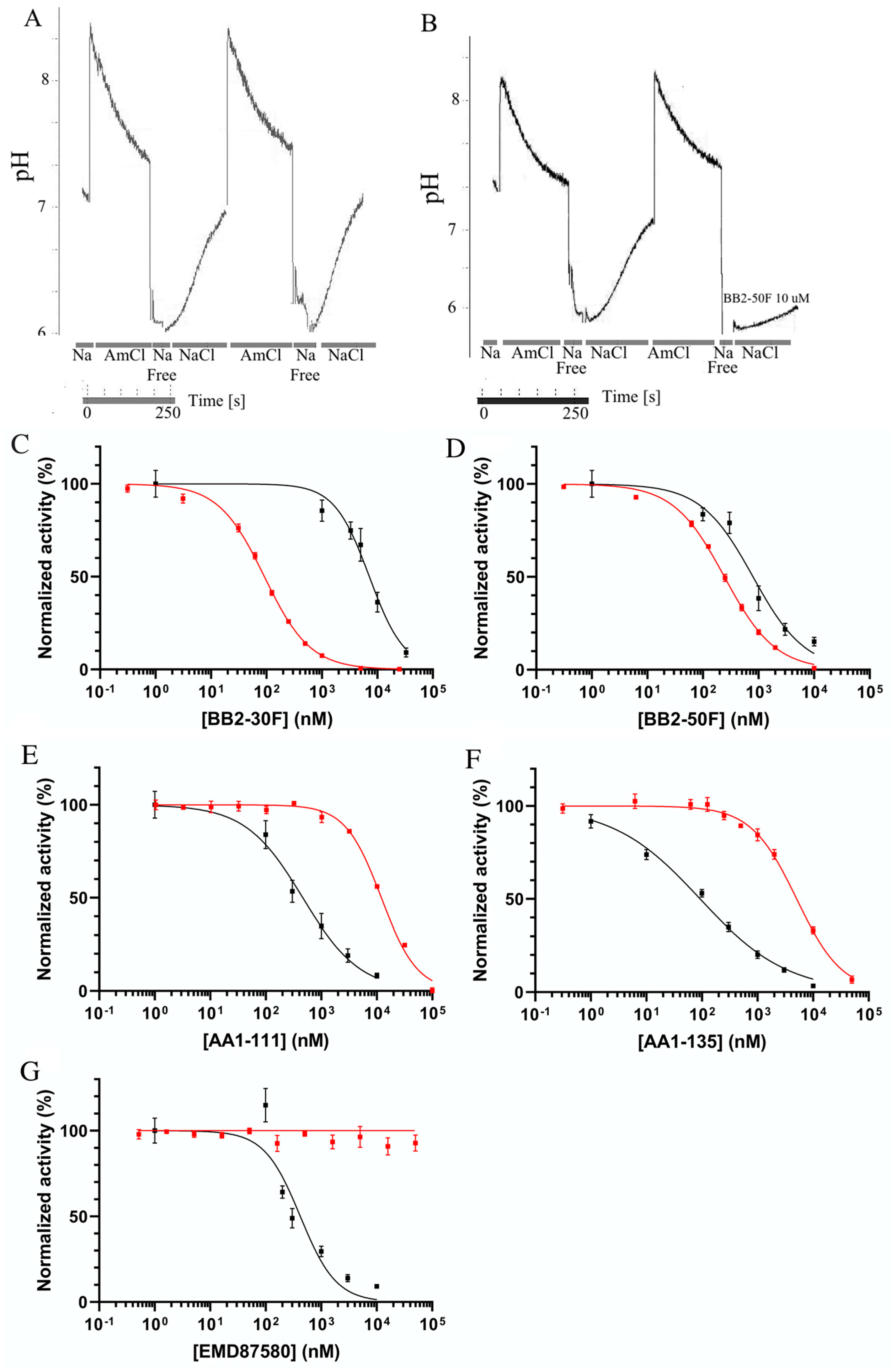
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Knockout (KO)/Reintroduction (RI) of NHE1
4.3. SDS-PAGE and Immunoblotting
4.4. Measurement of Intracellular pH
4.5. Cell Migration Assay
4.6. Cell Growth Assays
4.7. Anchorage-Dependent Colony Formation
4.8. Statistics
4.9. Inhibitors
4.10. Ex Ovo Chick Embryo Culture
4.11. Embryo Image Acquisition and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauseef, J.T.; Henry, M.D. Epithelial-to-mesenchymal transition in prostate cancer: Paradigm or puzzle? Nat. Rev. Urol. 2011, 8, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Dykes, S.S.; Gao, C.; Songock, W.K.; Bigelow, R.L.; Woude, G.V.; Bodily, J.M.; Cardelli, J.A. Zinc finger E-box binding homeobox-1 (Zeb1) drives anterograde lysosome trafficking and tumor cell invasion via upregulation of Na+/H+ Exchanger-1 (NHE1). Mol. Carcinog. 2017, 56, 722–734. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef]
- Andersen, A.P.; Moreira, J.M.; Pedersen, S.F. Interactions of ion transporters and channels with cancer cell metabolism and the tumour microenvironment. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130098. [Google Scholar] [CrossRef] [Green Version]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. pH control mechanisms of tumor survival and growth. J. Cell Physiol. 2011, 226, 299–308. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar]
- Greco, M.R.; Antelmi, E.; Busco, G.; Guerra, L.; Rubino, R.; Casavola, V.; Reshkin, S.J.; Cardone, R.A. Protease activity at invadopodial focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol. Rep. 2014, 31, 940–946. [Google Scholar] [CrossRef]
- Harguindey, S.; Stanciu, D.; Devesa, J.; Alfarouk, K.; Cardone, R.A.; Polo Orozco, J.D.; Devesa, P.; Rauch, C.; Orive, G.; Anitua, E.; et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 2017, 43, 157–179. [Google Scholar] [CrossRef]
- Amith, S.R.; Fliegel, L. Regulation of the Na+/H+ Exchanger (NHE1) in Breast Cancer Metastasis. Cancer Res. 2013, 73, 1259–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshkin, S.J.; Cardone, R.A.; Harguindey, S. Na+-H+ exchanger, pH regulation and cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 85–99. [Google Scholar] [CrossRef]
- Amith, S.R.; Wilkinson, J.M.; Baksh, S.; Fliegel, L. Na+/H+ exchanger (NHE1) as a novel co-adjuvant target in paclitaxel therapy of triple-negative breast cancer cells. Oncotarget 2015, 6, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Denker, S.P.; Barber, D.L. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J. Cell Biol. 2002, 159, 1087–1096. [Google Scholar] [CrossRef]
- Schneider, L.; Stock, C.M.; Dieterich, P.; Jensen, B.H.; Pedersen, L.B.; Satir, P.; Schwab, A.; Christensen, S.T.; Pedersen, S.F. The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR-alpha in the primary cilium. J. Cell Biol. 2009, 185, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amith, S.R.; Fong, S.; Baksh, S.; Fliegel, L. Na(+)/H(+) exchange in the tumour microenvironment: Does NHE1 drive breast cancer carcinogenesis? Int. J. Dev. Biol. 2015, 59, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, H.F.; Kwok, W.K.; Chan, K.K.; Chua, C.W.; Chan, Y.P.; Chu, Y.Y.; Wong, Y.C.; Wang, X.; Chan, K.W. TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis 2008, 29, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Kaur, P.; Tsao-Wei, D.; Naritoku, W.Y.; Groshen, S.; Datar, R.H.; Jones, L.W.; Imam, S.A. Unmethylated E-cadherin gene expression is significantly associated with metastatic human prostate cancer cells in bone. Prostate 2008, 68, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol. 2018, 1095, 101–110. [Google Scholar]
- Steffan, J.J.; Williams, B.C.; Welbourne, T.; Cardelli, J.A. HGF-induced invasion by prostate tumor cells requires anterograde lysosome trafficking and activity of Na+-H+ exchangers. J. Cell Sci. 2010, 123, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Margheri, F.; Luciani, C.; Taddei, M.L.; Giannoni, E.; Laurenzana, A.; Biagioni, A.; Chilla, A.; Chiarugi, P.; Fibbi, G.; Del Rosso, M. The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget 2014, 5, 1538–1553. [Google Scholar] [CrossRef] [Green Version]
- Bohm, L.; Serafin, A.; Akudugu, J.; Fernandez, P.; van der Merwe, A.; Aziz, N.A. uPA/PAI-1 ratios distinguish benign prostatic hyperplasia and prostate cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 1221–1228. [Google Scholar] [CrossRef]
- Akudugu, J.; Serafin, A.; Bohm, L. Further evaluation of uPA and PAI-1 as biomarkers for prostatic diseases. J. Cancer Res. Clin. Oncol. 2015, 141, 627–631. [Google Scholar] [CrossRef]
- Lippert, S.; Berg, K.D.; Hoyer-Hansen, G.; Lund, I.K.; Iversen, P.; Christensen, I.J.; Brasso, K.; Roder, M.A. Copenhagen uPAR prostate cancer (CuPCa) database: Protocol and early results. Biomark. Med. 2016, 10, 209–216. [Google Scholar] [CrossRef]
- Jankun, J.; Keck, R.W.; Skrzypczak-Jankun, E.; Swiercz, R. Inhibitors of urokinase reduce size of prostate cancer xenografts in severe combined immunodeficient mice. Cancer Res. 1997, 57, 559–563. [Google Scholar]
- Matthews, H.; Ranson, M.; Kelso, M.J. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: An orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer 2011, 129, 2051–2061. [Google Scholar] [CrossRef]
- Buckley, B.J.; Aboelela, A.; Minaei, E.; Jiang, L.X.; Xu, Z.; Ali, U.; Fildes, K.; Cheung, C.Y.; Cook, S.M.; Johnson, D.C.; et al. 6-Substituted Hexamethylene Amiloride (HMA) Derivatives as Potent and Selective Inhibitors of the Human Urokinase Plasminogen Activator for Use in Cancer. J. Med. Chem. 2018, 61, 8299–8320. [Google Scholar] [CrossRef]
- Li, X.; Fliegel, L. A novel human mutation in the SLC9A1 gene results in abolition of Na+/H+ exchanger activity. PLoS ONE 2015, 10, e0119453. [Google Scholar] [CrossRef]
- Li, X.; Dutta, D.; Jung, M.; Zimmermann, R.; Fliegel, L. Amino Acids 563-566 of the Na(+)/H(+) Exchanger Isoform 1 C-Terminal Cytosolic Tail Prevent Protein Degradation and Stabilize Protein Expression and Activity. Int. J. Mol. Sci. 2020, 21, 1737. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Wilkinson, J.M.; Fliegel, L. Assessing Na+/H+ exchange and cell effector functionality in metastatic breast cancer. Biochim. Open 2016, 2, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Fliegel, L. Role of pH regulatory proteins and dysregulation of pH in prostate cancer. In Reviews of Physiology, Biochemistry and Pharmacology; Springer Science & Business Media: Berlin, Germany, 2020. [Google Scholar]
- Buckley, B.J.; Kumar, A.; Aboelela, A.; Bujaroski, R.S.; Li, X.; Majed, H.; Fliegel, L.; Ranson, M.; Kelso, M.J. Screening of 5- and 6-Substituted Amiloride Libraries Identifies Dual-uPA/NHE1 Active and Single Target-Selective Inhibitors. Int. J. Mol. Sci. 2021, 22, 2999. [Google Scholar] [CrossRef]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoletov, K.; Willetts, L.; Paproski, R.J.; Bond, D.J.; Raha, S.; Jovel, J.; Adam, B.; Robertson, A.E.; Wong, F.; Woolner, E.; et al. Quantitative in vivo whole genome motility screen reveals novel therapeutic targets to block cancer metastasis. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Ferro, M.; Lucarelli, G.; Crocetto, F.; Dolce, P.; Verde, A.; La Civita, E.; Zappavigna, S.; de Cobelli, O.; Di Lorenzo, G.; Facchini, B.A.; et al. First-line systemic therapy for metastatic castration-sensitive prostate cancer: An updated systematic review with novel findings. Crit. Rev. Oncol. Hematol. 2021, 157, 103198. [Google Scholar] [CrossRef]
- Alimirah, F.; Chen, J.; Basrawala, Z.; Xin, H.; Choubey, D. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: Implications for the androgen receptor functions and regulation. FEBS Lett. 2006, 580, 2294–2300. [Google Scholar] [CrossRef] [Green Version]
- Harguindey, S.; Koltai, T.; Reshkin, S.J. Curing cancer? Further along the new pH-centric road and paradigm. Oncoscience 2018, 5, 132–133. [Google Scholar] [CrossRef]
- Koltai, T. Triple-edged therapy targeting intracellular alkalosis and extracellular acidosis in cancer. Semin. Cancer Biol. 2017, 43, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.J.; Snider, J.L.; Skalli, O.; Welbourne, T.; Cardelli, J.A. Na+/H+ exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic 2009, 10, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Amith, S.R.; Wilkinson, J.M.; Fliegel, L. Na+/H+ exchanger NHE1 regulation modulates metastatic potential and epithelial-mesenchymal transition of triple-negative breast cancer cells. Oncotarget 2016, 7, 21091–21113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegel, L. Structural and Functional Changes in the Na(+)/H(+) Exchanger Isoform 1, Induced by Erk1/2 Phosphorylation. Int. J. Mol. Sci. 2019, 20, 2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendus-Altenburger, R.; Pedraz-Cuesta, E.; Olesen, C.W.; Papaleo, E.; Schnell, J.A.; Hopper, J.T.; Robinson, C.V.; Pedersen, S.F.; Kragelund, B.B. The human Na(+)/H(+) exchanger 1 is a membrane scaffold protein for extracellular signal-regulated kinase 2. BMC Biol. 2016, 14, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karydis, A.; Jimenez-Vidal, M.; Denker, S.P.; Barber, D.L. Mislocalized scaffolding by the Na-H exchanger NHE1 dominantly inhibits fibronectin production and TGF-beta activation. Mol. Biol. Cell 2009, 20, 2327–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, S.; Morihara, H.; Yokoe, S.; Nakagawa, T.; Moriwaki, K.; Tomoda, K.; Asahi, M. Overexpression of Na(+)/H(+) exchanger 1 specifically induces cell death in human iPS cells via sustained activation of the Rho kinase ROCK. J. Biol. Chem. 2019, 294, 19577–19588. [Google Scholar] [CrossRef] [PubMed]
- Denker, S.P.; Huang, D.C.; Orlowski, J.; Furthmayr, H.; Barber, D.L. Direct binding of the Na-H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H(+) translocation. Mol. Cell 2000, 6, 1425–1436. [Google Scholar] [CrossRef]
- Wakabayashi, S.; Ikeda, T.; Iwamoto, T.; Pouyssegur, J.; Shigekawa, M. Calmodulin-binding autoinhibitory domain controls “pH-sensing” in the Na+/H+ exchanger NHE1 through sequence-specific interaction. Biochemistry 1997, 36, 12854–12861. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Su, X.; Wakabayashi, S.; Shigekawa, M. Calcineurin homologous protein as an essential cofactor for Na+/H+ exchangers. J. Biol. Chem. 2001, 276, 17367–17372. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, Y.; Alvarez, B.V.; Casey, J.R.; Fliegel, L. A novel carbonic anhydrase II binding site regulates NHE1 activity. Biochemistry 2006, 45, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; Litman, P.; Huang, L.; Ichimaru, E.; Furthmayr, H. Disruption of dynamic cell surface architecture of NIH3T3 fibroblasts by the N-terminal domains of moesin and ezrin: In vivo imaging with GFP fusion proteins. J. Cell Sci. 1999, 112 Pt 1, 111–125. [Google Scholar] [CrossRef]
- Martin, M.; Andreoli, C.; Sahuquet, A.; Montcourrier, P.; Algrain, M.; Mangeat, P. Ezrin NH2-terminal domain inhibits the cell extension activity of the COOH-terminal domain. J. Cell Biol. 1995, 128, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Jensen, H.H.; Pedersen, G.A.; Morgen, J.J.; Parsons, M.; Pedersen, S.F.; Nejsum, L.N. The Na(+)/H(+) exchanger NHE1 localizes as clusters to cryptic lamellipodia and accelerates collective epithelial cell migration. J. Physiol. 2019, 597, 849–867. [Google Scholar] [CrossRef] [Green Version]
- Hofschroer, V.; Koch, K.A.; Ludwig, F.T.; Friedl, P.; Oberleithner, H.; Stock, C.; Schwab, A. Extracellular protonation modulates cell-cell interaction mechanics and tissue invasion in human melanoma cells. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Buckley, B.J.; Majed, H.; Aboelela, A.; Minaei, E.; Jiang, L.; Fildes, K.; Cheung, C.Y.; Johnson, D.; Bachovchin, D.; Cook, G.M.; et al. 6-Substituted amiloride derivatives as inhibitors of the urokinase-type plasminogen activator for use in metastatic disease. Bioorg. Med. Chem. Lett. 2019, 29, 126753. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Jiang, Z.F.; Li, Q.X.; Liu, J.Y.; Wang, T.; Zhang, R.; Zhao, J.; Xu, Y.M.; Bao, W.; Zhang, Y.; et al. Inhibition of human breast cancer cell invasion by siRNA against urokinase-type plasminogen activator. Cancer Investig. 2010, 28, 689–697. [Google Scholar] [CrossRef]
- Bao, Y.N.; Cao, X.; Luo, D.H.; Sun, R.; Peng, L.X.; Wang, L.; Yan, Y.P.; Zheng, L.S.; Xie, P.; Cao, Y.; et al. Urokinase-type plasminogen activator receptor signaling is critical in nasopharyngeal carcinoma cell growth and metastasis. Cell Cycle 2014, 13, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ling, D.; Tan, J.; Zhang, J.; Li, L. Expression of urokinase plasminogen activator and plasminogen activator inhibitor type-1 in ovarian cancer and its clinical significance. Oncol. Rep. 2013, 29, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, C.A.; Scott, L.J.; Bagley, S.; Bryden, A.A.; Clarke, N.W.; Lang, S.H. Role of proteolytic enzymes in human prostate bone metastasis formation: In vivo and in vitro studies. Br. J. Cancer 2002, 86, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cozzi, P.J. Targeting uPA/uPAR in prostate cancer. Cancer Treat. Rev. 2007, 33, 521–527. [Google Scholar] [CrossRef]
- Li, X.; Ding, J.; Liu, Y.; Brix, B.J.; Fliegel, L. Functional analysis of acidic amino acids in the cytosolic tail of the Na+/H+ exchanger. Biochemistry 2004, 43, 16477–16486. [Google Scholar] [CrossRef]
- Ding, J.; Rainey, J.K.; Xu, C.; Sykes, B.D.; Fliegel, L. Structural and functional characterization of transmembrane segment VII of the Na+/H+ exchanger isoform 1. J. Biol. Chem. 2006, 281, 29817–29829. [Google Scholar] [CrossRef] [Green Version]
- Guffey, S.C.; Fliegel, L.; Goss, G.G. Cloning and characterization of Na(+)/H(+) Exchanger isoforms NHE2 and NHE3 from the gill of Pacific dogfish Squalus suckleyi. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2015, 188, 46–53. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BB2-30F | BB2-50F | AA1-111 | AA1-135 | EMD87580 |
|---|---|---|---|---|---|
| NHE1 | 7.2 | 0.79 | 0.45 | 0.09 | 0.46 |
| uPA | 0.09 | 0.24 | 12.1 | 5.1 | N/A |
| Name | Guide Sequence | Target Site |
|---|---|---|
| NHE1CRISPR1f | caccgGATCAACAACATCGGCCTCC | Exon 2 |
| NHE1CRISPR1r | aaacGGAGGCCGATGTTGTTGATC | |
| NHE1CRISPR2f | caccgCGGGACGATGCTTGAGATAG | Exon 2 |
| NHE1CRISPR2r | aaacCTATCTCAAGCATCGTCCCG | |
| NHE1CRISPR3f | caccgGTTTGCCAACTACGAACACG | Exon 3 |
| NHE1CRISPR3r | aaacCGTGTTCGTAGTTGGCAAAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Buckley, B.; Stoletov, K.; Jing, Y.; Ranson, M.; Lewis, J.D.; Kelso, M.; Fliegel, L. Roles of the Na+/H+ Exchanger Isoform 1 and Urokinase in Prostate Cancer Cell Migration and Invasion. Int. J. Mol. Sci. 2021, 22, 13263. https://doi.org/10.3390/ijms222413263
Li X, Buckley B, Stoletov K, Jing Y, Ranson M, Lewis JD, Kelso M, Fliegel L. Roles of the Na+/H+ Exchanger Isoform 1 and Urokinase in Prostate Cancer Cell Migration and Invasion. International Journal of Molecular Sciences. 2021; 22(24):13263. https://doi.org/10.3390/ijms222413263
Chicago/Turabian StyleLi, Xiuju, Benjamin Buckley, Konstantin Stoletov, Yang Jing, Marie Ranson, John D. Lewis, Mike Kelso, and Larry Fliegel. 2021. "Roles of the Na+/H+ Exchanger Isoform 1 and Urokinase in Prostate Cancer Cell Migration and Invasion" International Journal of Molecular Sciences 22, no. 24: 13263. https://doi.org/10.3390/ijms222413263