
Adipose Lipolysis Regulates Cardiac Glucose Uptake and Function in Mice under Cold Stress



 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
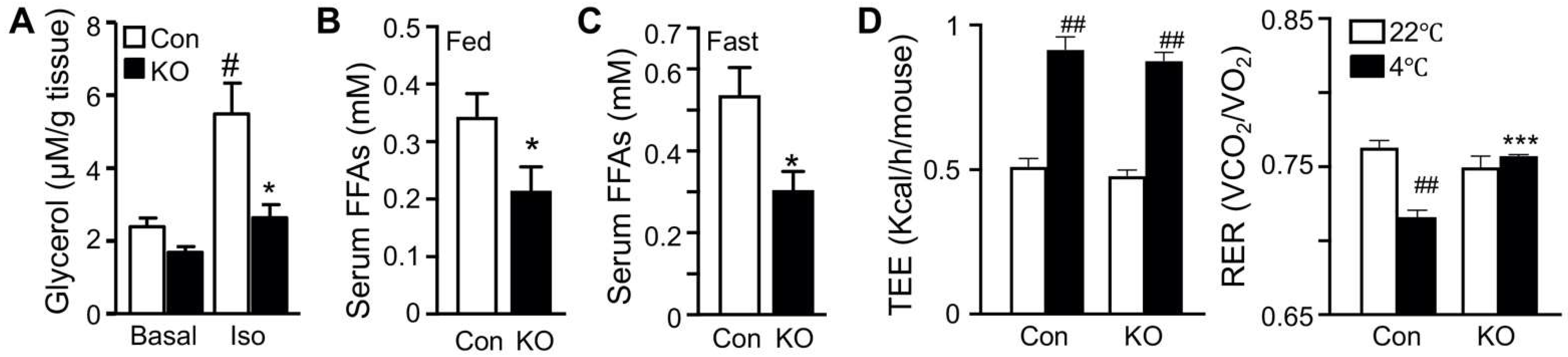
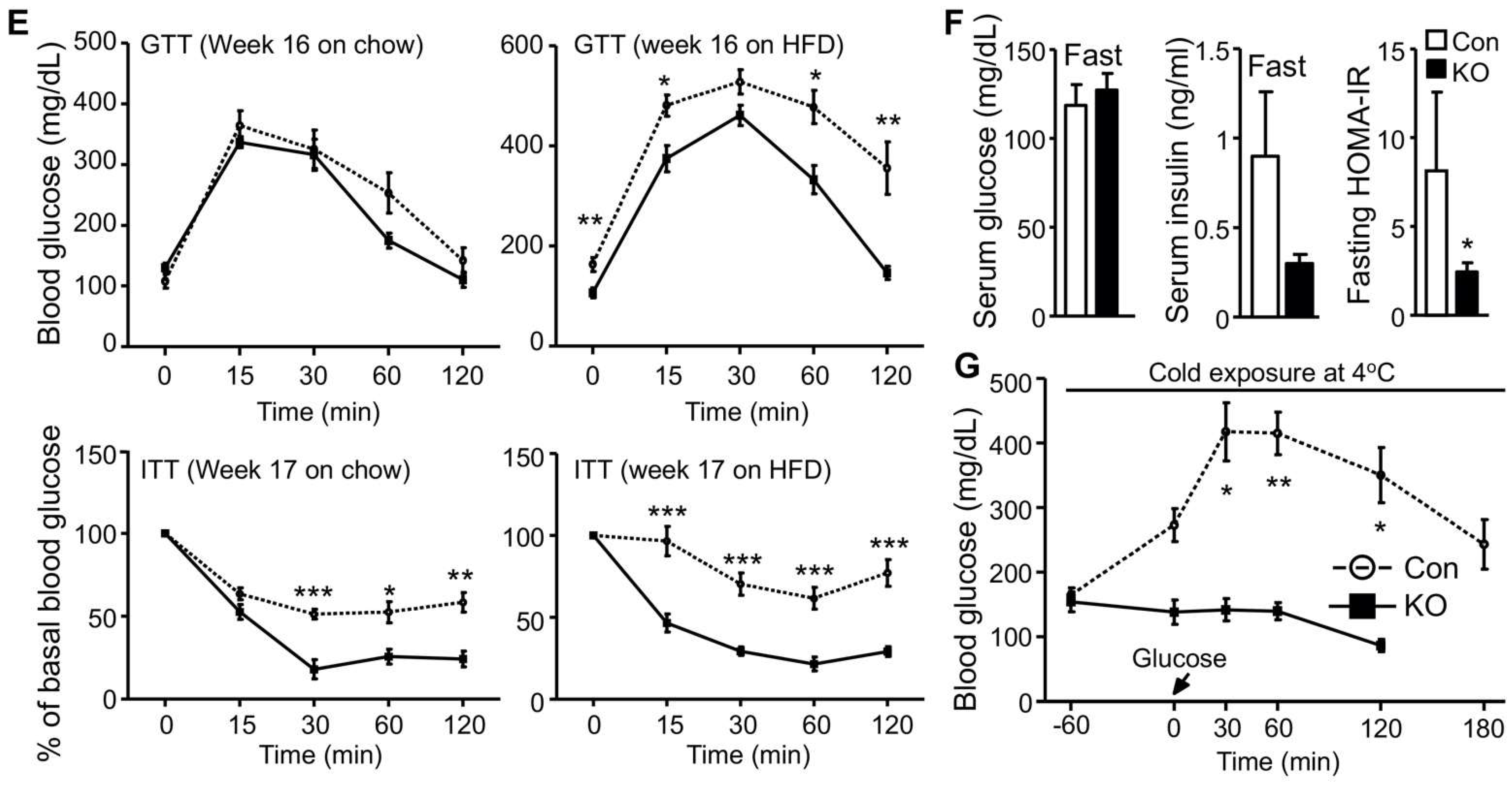
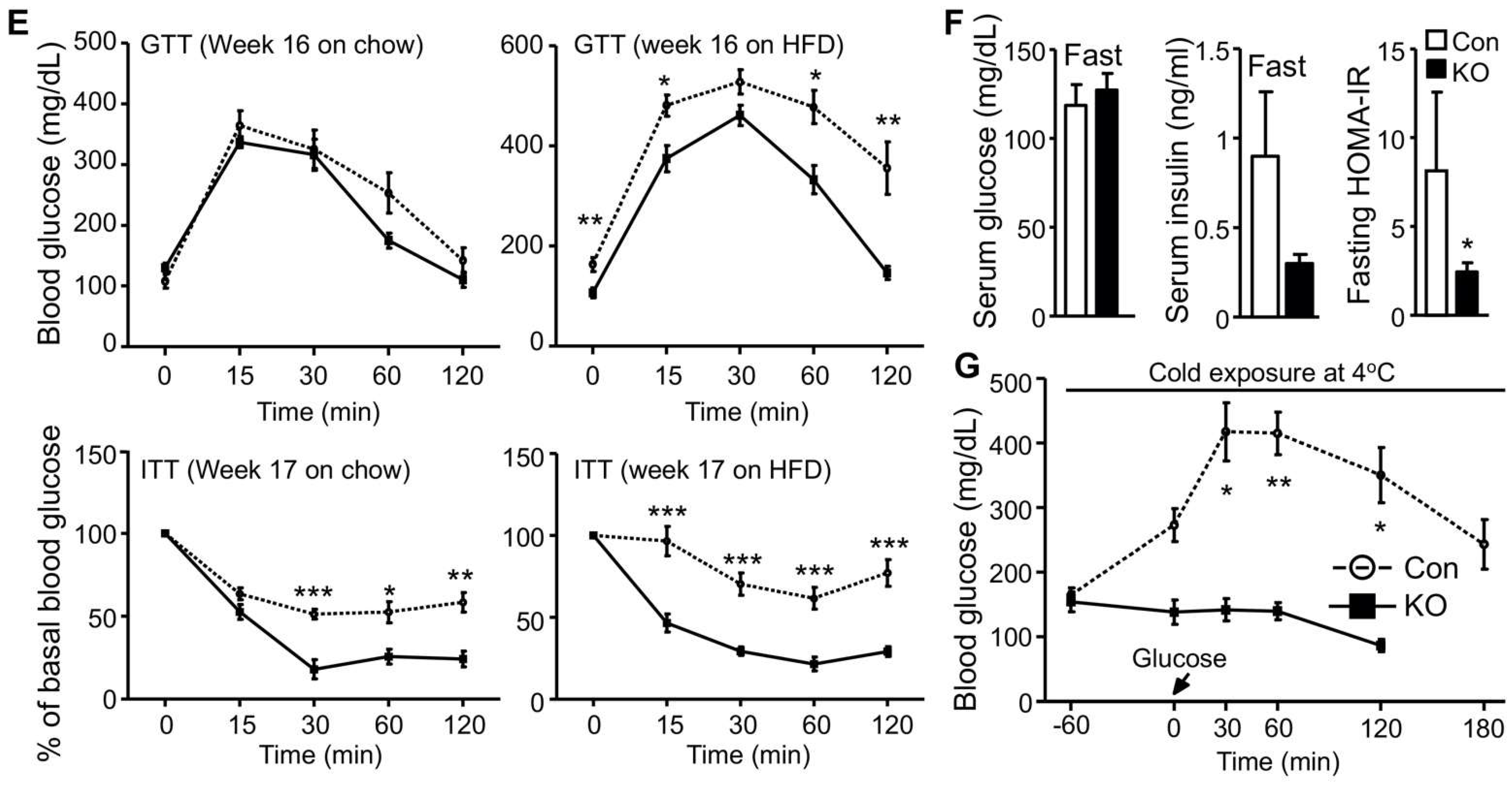
2.1. Selective Inactivation of CGI-58 in Adipose Tissue Reduces Blood Free Fatty Acids and Increases Whole-Body Glucose Utilization
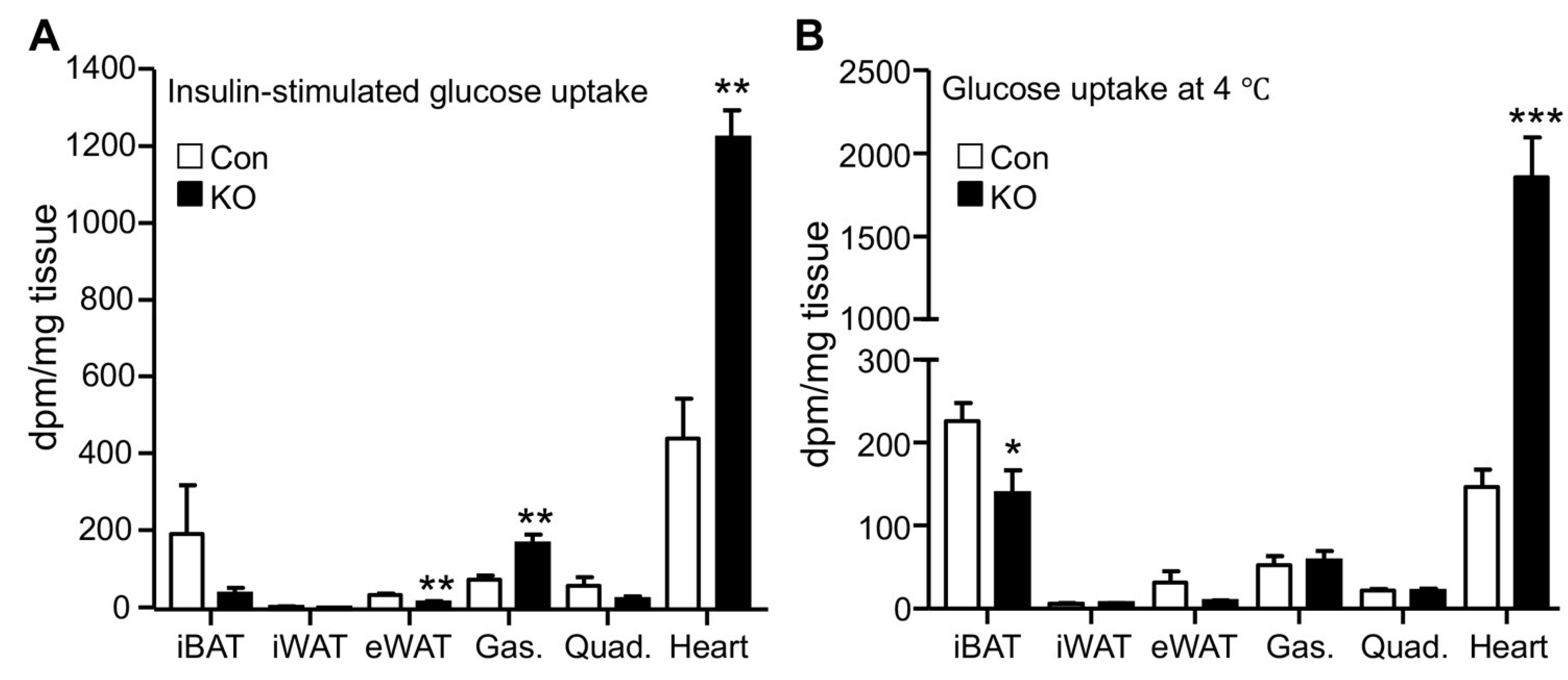
2.2. CGI-58 Deletion in the Adipose Tissue Increases Glucose Uptake in the Heart
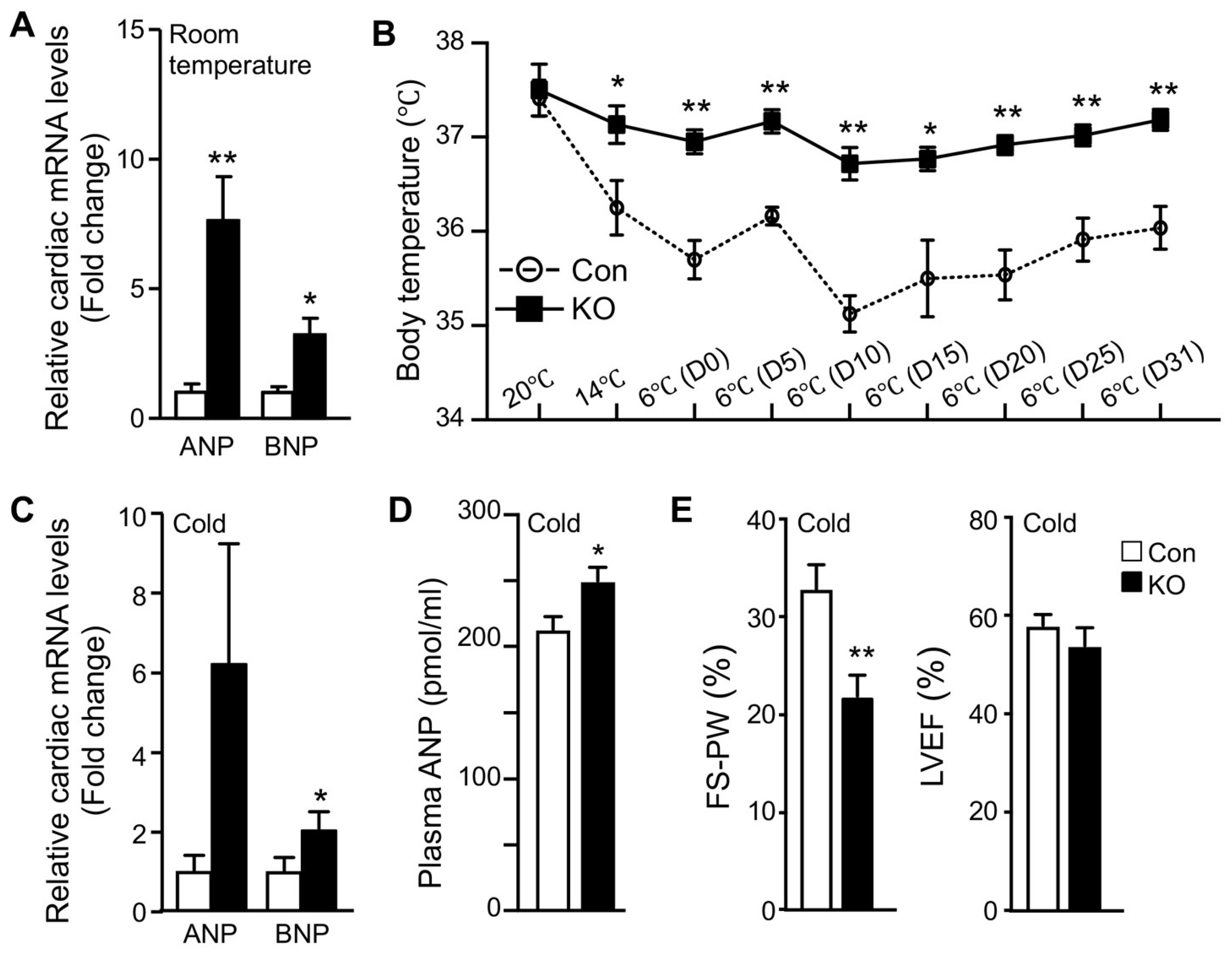
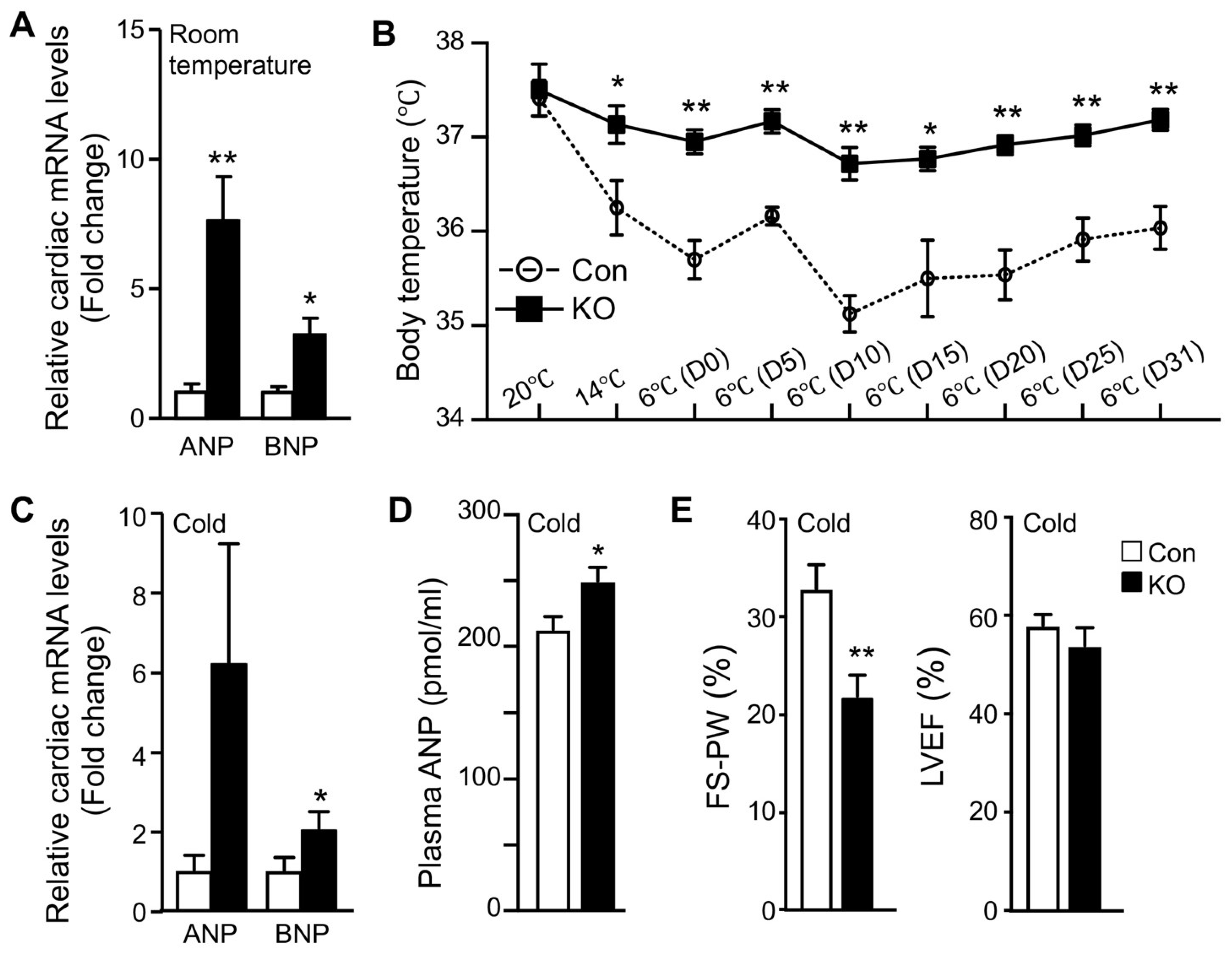
2.3. Adipose CGI-58 Deficiency Increases Cardiac Expression of Natriuretic Peptides
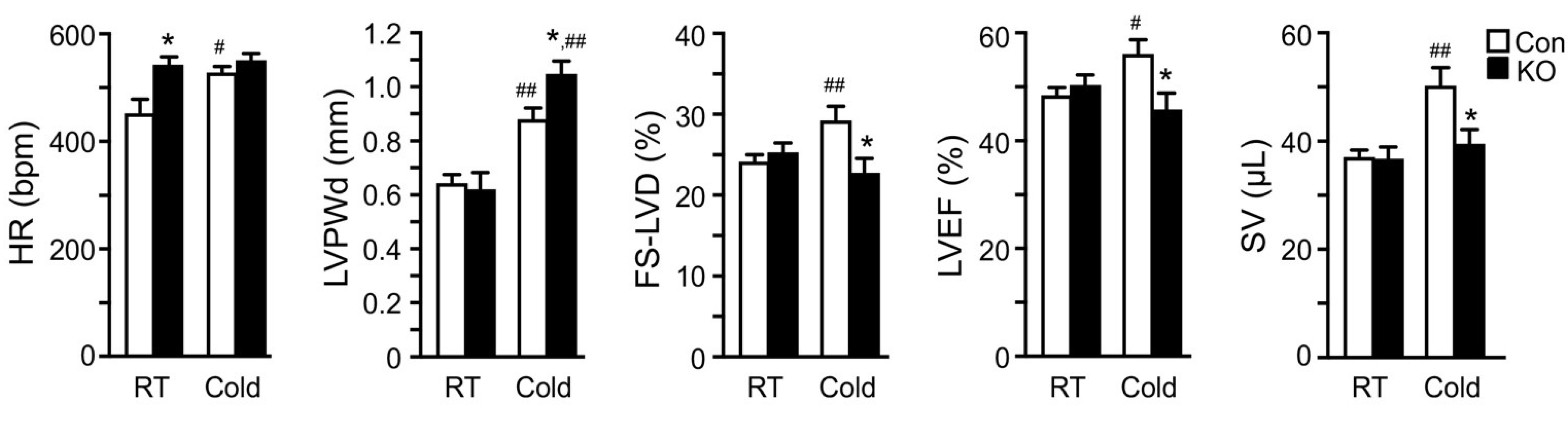
2.4. Adipose CGI-58 Deficiency Attenuates Cardiac Function during Cold Exposure
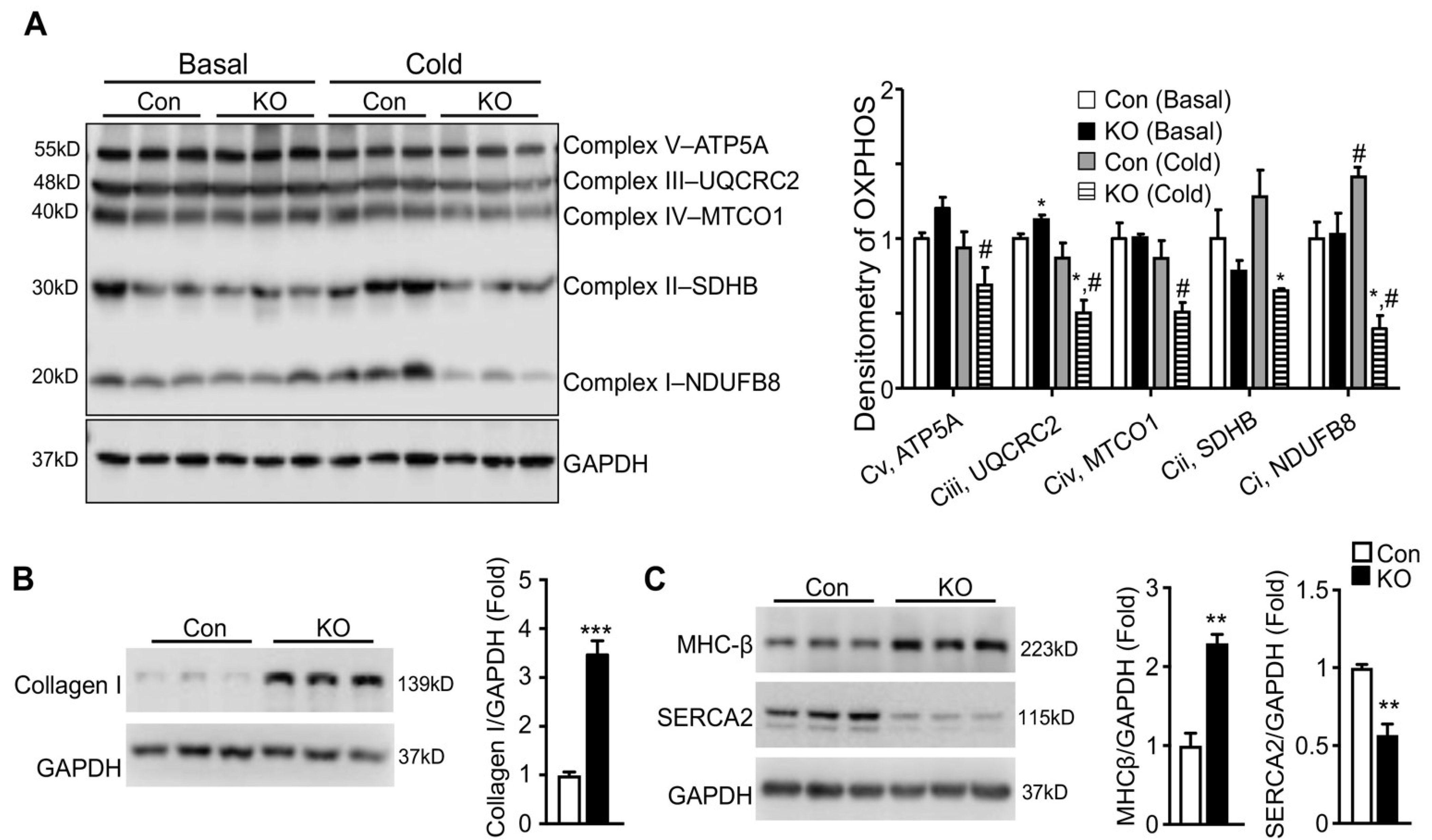
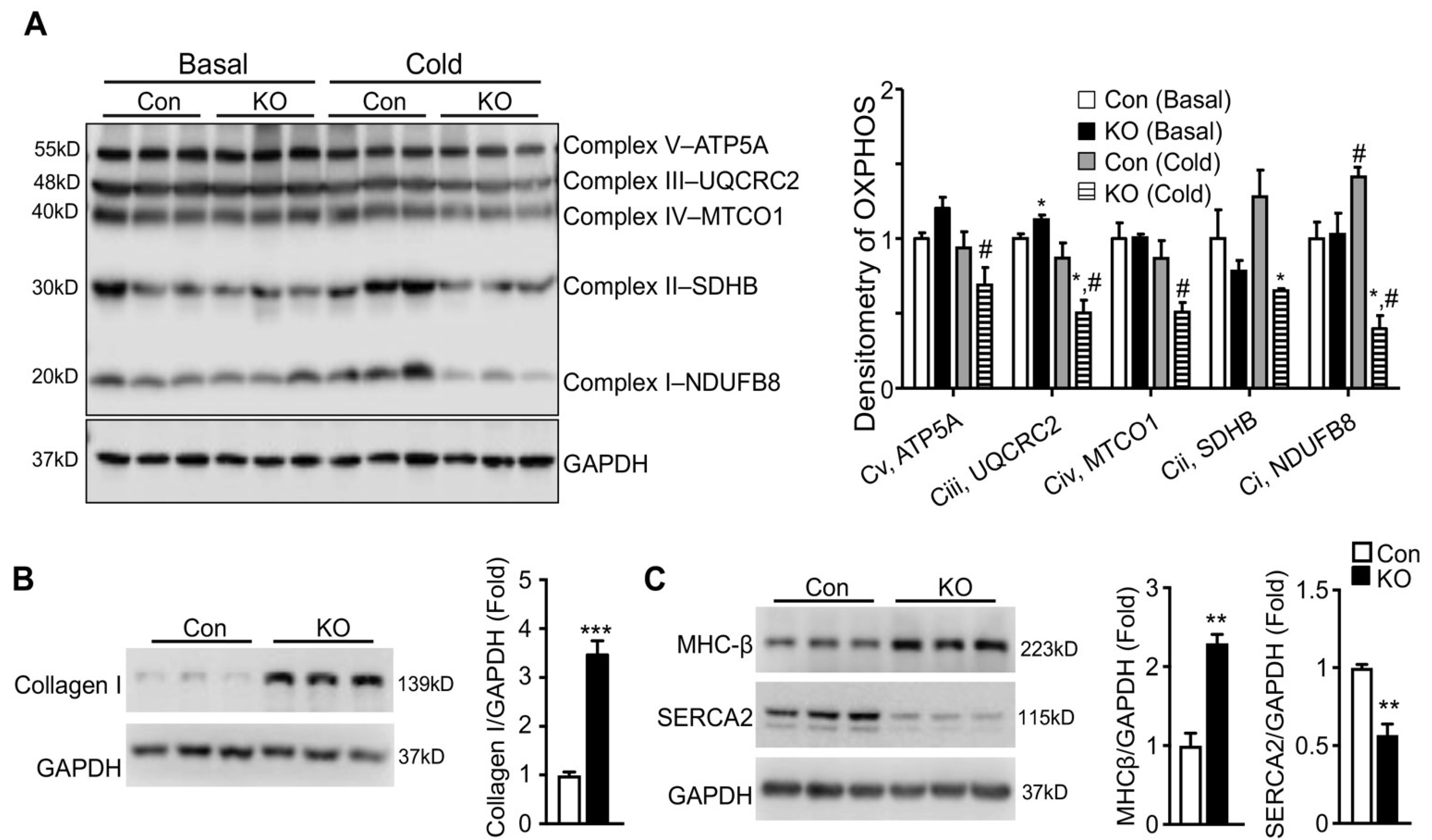
2.5. Adipose CGI-58 Deficiency Induces Pathological Remodeling of the Heart
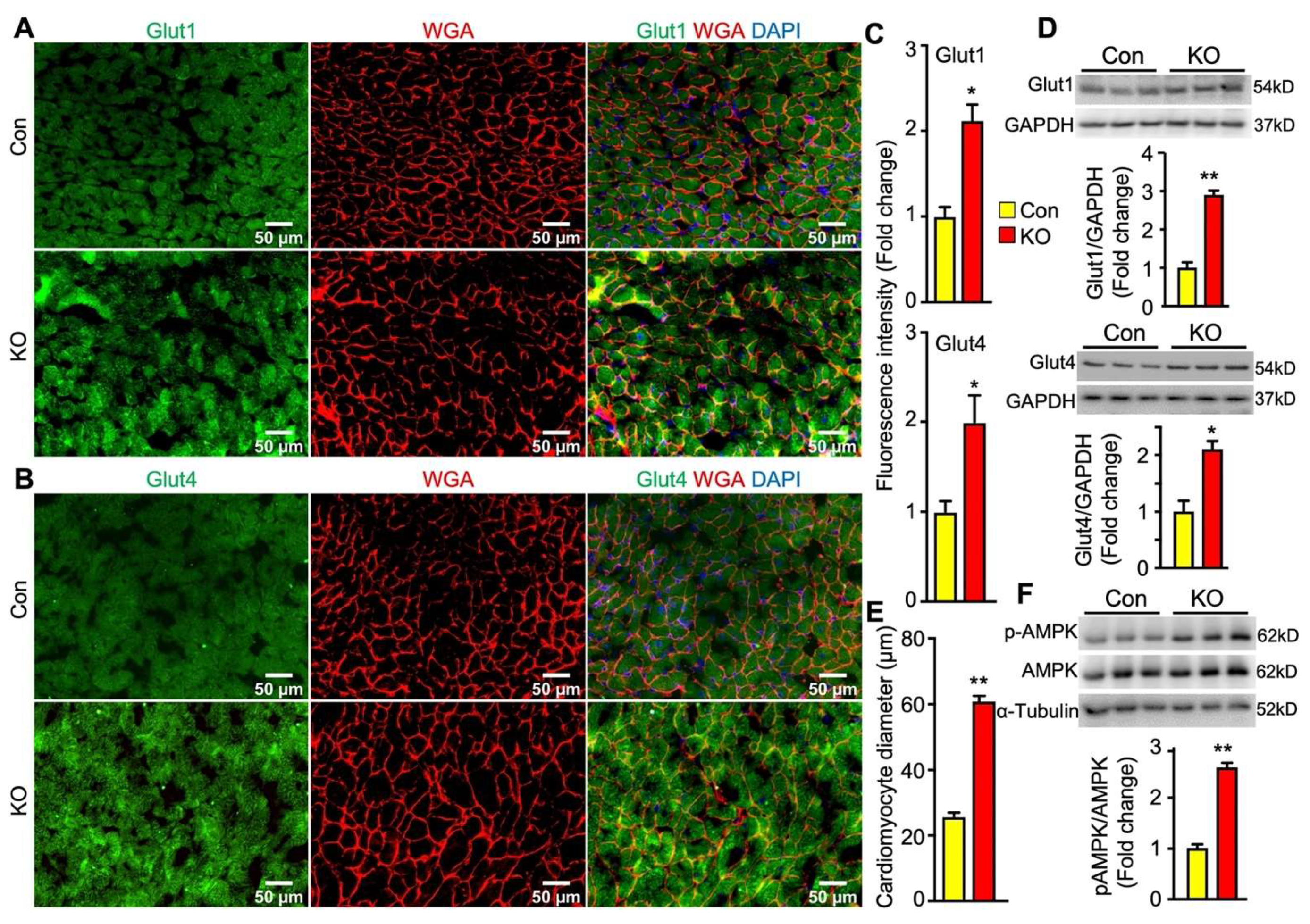
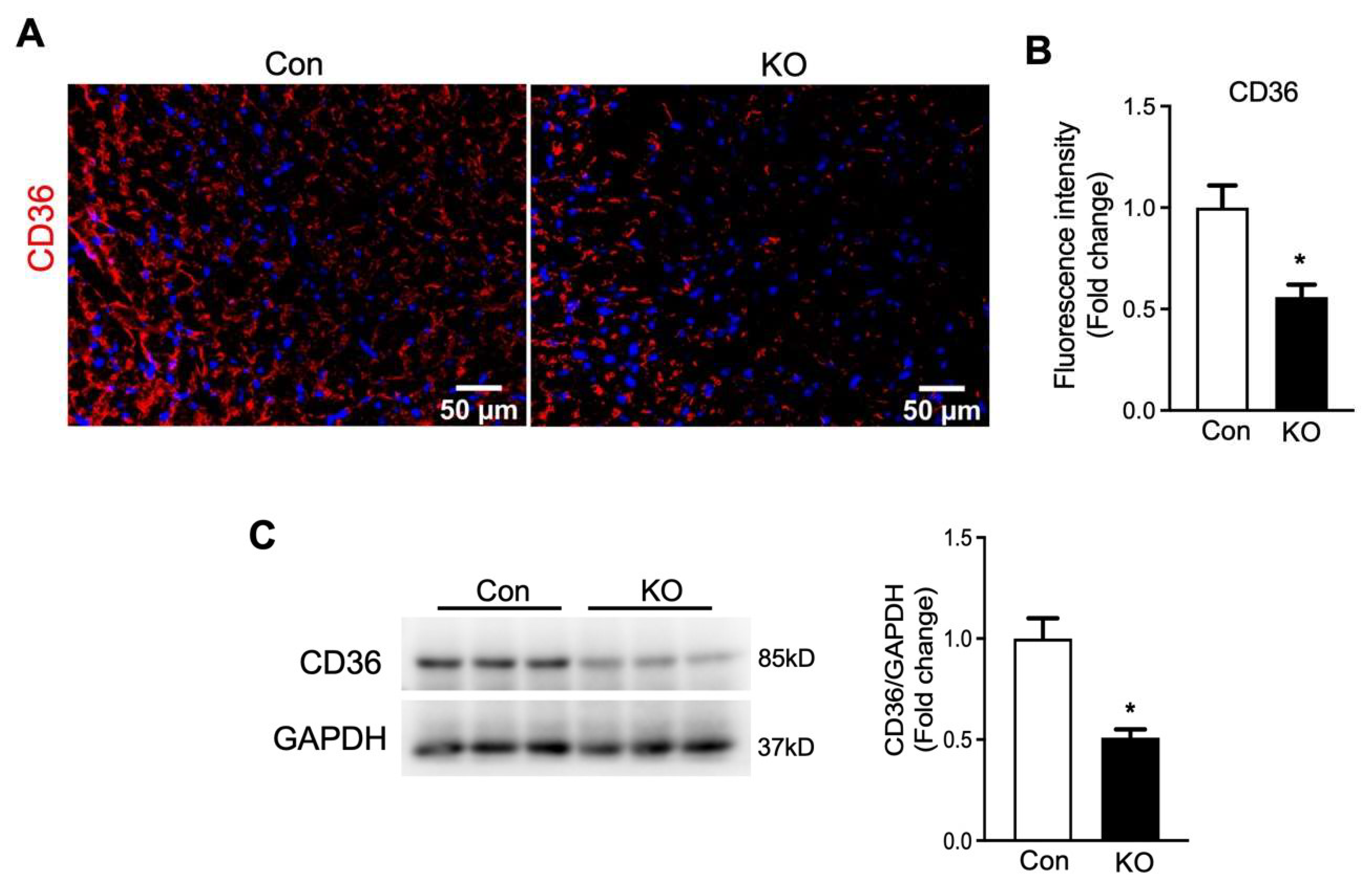
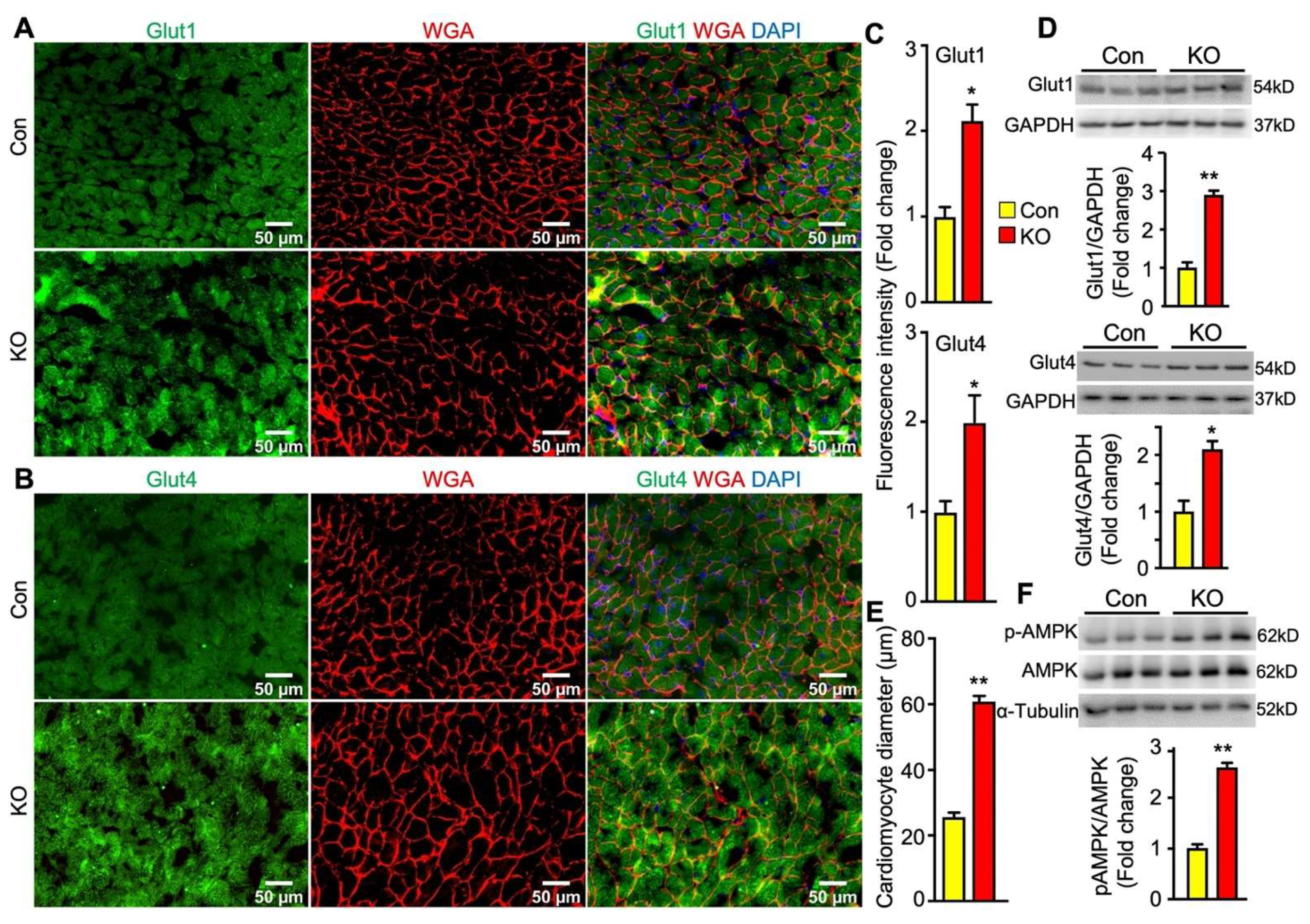
2.6. Adipose CGI-58 Deficiency Increases Glucose Transporter Expression Levels and Cardiomyocytes in Hearts of Chow-Fed Mice after Chronic Cold Stress
3. Discussion
4. Materials and Methods
4.1. Animals and Diets
4.2. Indirect Calorimetry
4.3. Glucose and Insulin Tolerance Tests
4.4. Blood Analysis
4.5. Tissue Glucose Uptake Assay
4.6. Gene Expression Analysis
4.7. Echocardiography
4.8. Immunoblotting
4.9. Immunofluorescent Staining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.G.; Zechner, R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 2013, 27, 459–484. [Google Scholar] [CrossRef] [Green Version]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.L.; Farese, R.V., Jr. Fat breakdown: A function for CGI-58 (ABHD5) provides a new piece of the puzzle. Cell Metab. 2006, 3, 305–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Ma, Y.; Chanturiya, T.; Cao, Q.; Wang, Y.; Kadegowda, A.K.G.; Jackson, R.; Rumore, D.; Xue, B.; Shi, H.; et al. Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell Metab. 2017, 26, 764–777.e765. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Shi, H.; Xue, B.; Yu, L. What activates thermogenesis when lipid droplet lipolysis is absent in brown adipocytes? Adipocyte 2018, 27, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.; Diwoky, C.; Schoiswohl, G.; Feiler, U.; Wongsiriroj, N.; Abdellatif, M.; Kolb, D.; Hoeks, J.; Kershaw, E.E.; Sedej, S.; et al. Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab. 2017, 26, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Fuster, J.J.; Walsh, K. Adipokines: A link between obesity and cardiovascular disease. J. Cardiol. 2014, 63, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Akoumianakis, I.; Antoniades, C. The interplay between adipose tissue and the cardiovascular system: Is fat always bad? Cardiovasc. Res. 2017, 113, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salatzki, J.; Foryst-Ludwig, A.; Bentele, K.; Blumrich, A.; Smeir, E.; Ban, Z.; Brix, S.; Grune, J.; Beyhoff, N.; Klopfleisch, R.; et al. Adipose tissue ATGL modifies the cardiac lipidome in pressure-overload-induced left ventricular failure. PLoS Genet. 2018, 14, e1007171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foryst-Ludwig, A.; Kreissl, M.C.; Benz, V.; Brix, S.; Smeir, E.; Ban, Z.; Januszewicz, E.; Salatzki, J.; Grune, J.; Schwanstecher, A.K.; et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J. Biol. Chem. 2015, 290, 23603–23615. [Google Scholar] [CrossRef] [Green Version]
- Ikaheimo, T.M. Cardiovascular diseases, cold exposure and exercise. Temperature 2018, 5, 123–146. [Google Scholar] [CrossRef] [PubMed]
- Bordicchia, M.; Liu, D.; Amri, E.Z.; Ailhaud, G.; Dessi-Fulgheri, P.; Zhang, C.; Takahashi, N.; Sarzani, R.; Collins, S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest. 2012, 122, 1022–1036. [Google Scholar] [CrossRef] [Green Version]
- Carper, D.; Coue, M.; Nascimento, E.B.M.; Barquissau, V.; Lagarde, D.; Pestourie, C.; Laurens, C.; Petit, J.V.; Soty, M.; Monbrun, L.; et al. Atrial Natriuretic Peptide Orchestrates a Coordinated Physiological Response to Fuel Non-shivering Thermogenesis. Cell Rep. 2020, 32, 108075. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Sergeeva, I.A.; Christoffels, V.M. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim. Biophys Acta 2013, 1832, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessin, J.E.; Bell, G.I. Mammalian facilitative glucose transporter family: Structure and molecular regulation. Annu. Rev. Physiol. 1992, 54, 911–930. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Till, M.; Ouwens, D.M.; Kessler, A.; Eckel, J. Molecular mechanisms of contraction-regulated cardiac glucose transport. Biochem. J. 2000, 346, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., III; Bergeron, R.; Shulman, G.I.; Young, L.H. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol. 1999, 277, H643–H649. [Google Scholar] [CrossRef] [PubMed]
- Tuunanen, H.; Engblom, E.; Naum, A.; Nagren, K.; Hesse, B.; Airaksinen, K.E.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006, 114, 2130–2137. [Google Scholar] [CrossRef] [Green Version]
- Blachere, J.C.; Perusse, F.; Bukowiecki, L.J. Lowering plasma free fatty acids with Acipimox mimics the antidiabetic effects of the beta 3-adrenergic agonist CL-316243 in obese Zucker diabetic fatty rats. Metabolism 2001, 50, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B. Reducing plasma free fatty acids by acipimox improves glucose tolerance in high-fat fed mice. Acta Physiol. Scand. 2001, 171, 161–167. [Google Scholar] [CrossRef]
- Knuuti, M.J.; Maki, M.; Yki-Jarvinen, H.; Voipio-Pulkki, L.M.; Harkonen, R.; Haaparanta, M.; Nuutila, P. The effect of insulin and FFA on myocardial glucose uptake. J. Mol. Cell Cardiol. 1995, 27, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Pascual, F.; Coleman, R.A. Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim. Biophys Acta 2016, 1861, 1425–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.T.; Ussher, J.R.; Mohammad, A.; Lam, A.; Lopaschuk, G.D. 5’-AMP-activated protein kinase increases glucose uptake independent of GLUT4 translocation in cardiac myocytes. Can. J. Physiol. Pharmacol. 2014, 92, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. An Emerging Role of Natriuretic Peptides: Igniting the Fat Furnace to Fuel and Warm the Heart. Mayo Clin. Proc. 2015, 90, 1666–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heine, M.; Fischer, A.W.; Schlein, C.; Jung, C.; Straub, L.G.; Gottschling, K.; Mangels, N.; Yuan, Y.; Nilsson, S.K.; Liebscher, G.; et al. Lipolysis Triggers a Systemic Insulin Response Essential for Efficient Energy Replenishment of Activated Brown Adipose Tissue in Mice. Cell Metab. 2018, 28, 644–655.e644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouellet, V.; Labbe, S.M.; Blondin, D.P.; Phoenix, S.; Guerin, B.; Haman, F.; Turcotte, E.E.; Richard, D.; Carpentier, A.C. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Invest. 2012, 122, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.L.; Tseng, Y.H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Mottillo, E.P.; Balasubramanian, P.; Lee, Y.H.; Weng, C.; Kershaw, E.E.; Granneman, J.G. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic beta3-adrenergic receptor activation. J. Lipid Res. 2014, 55, 2276–2286. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.H.; Chang, J.S.; Jo, Y.H. Intracellular glycolysis in brown adipose tissue is essential for optogenetically induced nonshivering thermogenesis in mice. Sci. Rep. 2018, 8, 6672. [Google Scholar] [CrossRef]
- Wang, Z.; Ning, T.; Song, A.; Rutter, J.; Wang, Q.A.; Jiang, L. Chronic cold exposure enhances glucose oxidation in brown adipose tissue. EMBO Rep. 2020, 21, e50085. [Google Scholar] [CrossRef] [PubMed]
- Blondin, D.P.; Frisch, F.; Phoenix, S.; Guerin, B.; Turcotte, E.E.; Haman, F.; Richard, D.; Carpentier, A.C. Inhibition of Intracellular Triglyceride Lipolysis Suppresses Cold-Induced Brown Adipose Tissue Metabolism and Increases Shivering in Humans. Cell Metab. 2017, 25, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.L. Cardiac energetics. Physiol. Rev. 1978, 58, 174–254. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parajuli, N.; Takahara, S.; Matsumura, N.; Kim, T.T.; Ferdaoussi, M.; Migglautsch, A.K.; Zechner, R.; Breinbauer, R.; Kershaw, E.E.; Dyck, J.R.B. Atglistatin ameliorates functional decline in heart failure via adipocyte-specific inhibition of adipose triglyceride lipase. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H879–H884. [Google Scholar] [CrossRef] [PubMed]
- Halbirk, M.; Norrelund, H.; Moller, N.; Schmitz, O.; Gotzsche, L.; Nielsen, R.; Nielsen-Kudsk, J.E.; Nielsen, S.S.; Nielsen, T.T.; Eiskjaer, H.; et al. Suppression of circulating free fatty acids with acipimox in chronic heart failure patients changes whole body metabolism but does not affect cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1220–H1225. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoiswohl, G.; Stefanovic-Racic, M.; Menke, M.N.; Wills, R.C.; Surlow, B.A.; Basantani, M.K.; Sitnick, M.T.; Cai, L.; Yazbeck, C.F.; Stolz, D.B.; et al. Impact of Reduced ATGL-Mediated Adipocyte Lipolysis on Obesity-Associated Insulin Resistance and Inflammation in Male Mice. Endocrinology 2015, 156, 3610–3624. [Google Scholar] [CrossRef] [Green Version]
- Dube, J.J.; Sitnick, M.T.; Schoiswohl, G.; Wills, R.C.; Basantani, M.K.; Cai, L.; Pulinilkunnil, T.; Kershaw, E.E. Adipose triglyceride lipase deletion from adipocytes, but not skeletal myocytes, impairs acute exercise performance in mice. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E879–E890. [Google Scholar] [CrossRef]
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A.; et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Li, Y.; Grise, A.; Wang, H. CGI-58: Versatile Regulator of Intracellular Lipid Droplet Homeostasis. Adv. Exp. Med. Biol. 2020, 1276, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Readnower, R.D.; Salabei, J.K.; Long, B.W.; Aird, A.L.; Zheng, Y.T.; Muthusamy, S.; Facundo, H.T.; Hill, B.G.; Jones, S.P. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem. J. 2015, 467, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brower, G.L.; Gardner, J.D.; Forman, M.F.; Murray, D.B.; Voloshenyuk, T.; Levick, S.P.; Janicki, J.S. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur. J. Cardiothorac. Surg. 2006, 30, 604–610. [Google Scholar] [CrossRef]
- Harada, K.; Sugaya, T.; Murakami, K.; Yazaki, Y.; Komuro, I. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation 1999, 100, 2093–2099. [Google Scholar] [CrossRef]
- Jones, W.K.; Grupp, I.L.; Doetschman, T.; Grupp, G.; Osinska, H.; Hewett, T.E.; Boivin, G.; Gulick, J.; Ng, W.A.; Robbins, J. Ablation of the murine alpha myosin heavy chain gene leads to dosage effects and functional deficits in the heart. J. Clin. Invest. 1996, 98, 1906–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, A.; Simonides, W.S. Regulation of myocardial SERCA2a expression in ventricular hypertrophy and heart failure. Future Cardiol. 2005, 1, 543–553. [Google Scholar] [CrossRef]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, P.; Watson, C.J.; van Es, M.H.; Phelan, D.; McGorrian, C.; Tolan, M.; Ledwidge, M.T.; McDonald, K.M.; Baugh, J.A. Getting to the heart of cardiac remodeling; how collagen subtypes may contribute to phenotype. J. Mol. Cell Cardiol. 2012, 52, 148–153. [Google Scholar] [CrossRef]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.S.; Zhang, J.; Li, Y.F.; Chen, B.R.; Khurwolah, M.R.; Tian, Y.F.; Shi, H.J.; Yang, Z.J.; Wang, L.S. A pilot clinical study of adjunctive therapy with selective intracoronary hypothermia in patients with ST-segment elevation myocardial infarction. Catheter. Cardiovasc. Interv. 2018, 92, E433–E440. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, P.A.; Rao, G.; Briceno, D.F.; Lombardo, M.; Ramakrishna, H.; Bortnick, A.; Garcia, M.; Menegus, M.; Sims, D.; Makkiya, M.; et al. Therapeutic hypothermia in ST elevation myocardial infarction: A systematic review and meta-analysis of randomised control trials. Heart 2016, 102, 712–719. [Google Scholar] [CrossRef]
- Simkhovich, B.Z.; Hale, S.L.; Kloner, R.A. Metabolic mechanism by which mild regional hypothermia preserves ischemic tissue. J. Cardiovasc. Pharmacol. Ther. 2004, 9, 83–90. [Google Scholar] [CrossRef]
- Scherz, T.; Hofbauer, T.M.; Ondracek, A.S.; Simon, D.; Sterz, F.; Testori, C.; Lang, I.M.; Mangold, A. Mild Therapeutic Hypothermia Alters Hemostasis in ST Elevation Myocardial Infarction Patients. Front. Cardiovasc. Med. 2021, 8, 707367. [Google Scholar] [CrossRef] [PubMed]
- Otterspoor, L.C.; Van ’t Veer, M.; Van Nunen, L.X.; Brueren, G.R.G.; Tonino, P.A.L.; Wijnbergen, I.F.; Helmes, H.; Zimmermann, F.M.; Van Hagen, E.; Johnson, N.P.; et al. Safety and feasibility of selective intracoronary hypothermia in acute myocardial infarction. EuroIntervention 2017, 13, e1475–e1482. [Google Scholar] [CrossRef]
- Meerbaum, S.; Haendchen, R.V.; Corday, E.; Povzhitkov, M.; Fishbein, M.C.; J, Y.R.; Lang, T.W.; Uchiyama, T.; Aosaki, N.; Broffman, J. Hypothermic coronary venous phased retroperfusion: A closed-chest treatment of acute regional myocardial ischemia. Circulation 1982, 65, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Bashtawi, Y.; Almuwaqqat, Z. Therapeutic Hypothermia in STEMI. Cardiovasc. Revasc. Med. 2021, 29, 77–84. [Google Scholar] [CrossRef]
- Opie, L.H. Reperfusion injury and its pharmacologic modification. Circulation 1989, 80, 1049–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnieri, C.; Flamigni, F.; Caldarera, C.M. Role of oxygen in the cellular damage induced by re-oxygenation of hypoxic heart. J. Mol. Cell Cardiol. 1980, 12, 797–808. [Google Scholar] [CrossRef]
- Kim, J.K.; Gavrilova, O.; Chen, Y.; Reitman, M.L.; Shulman, G.I. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J. Biol. Chem 2000, 275, 8456–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, J.; Hu, X.; Philipson, K.D.; Scharf, S.M. The Na+/Ca2+ exchanger-1 mediates left ventricular dysfunction in mice with chronic intermittent hypoxia. J. Appl. Physiol. 2010, 109, 1675–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Einbinder, E.; Zhang, Q.; Hasday, J.; Balke, C.W.; Scharf, S.M. Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. Am. J. Respir. Crit. Care Med. 2005, 172, 915–920. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.; Shin, H.; Tang, Z.; Yeh, Y.; Ma, Y.; Kadegowda, A.K.G.; Wang, H.; Jiang, L.; Arya, R.K.; Chen, L.; et al. Adipose Lipolysis Regulates Cardiac Glucose Uptake and Function in Mice under Cold Stress. Int. J. Mol. Sci. 2021, 22, 13361. https://doi.org/10.3390/ijms222413361
Choi Y, Shin H, Tang Z, Yeh Y, Ma Y, Kadegowda AKG, Wang H, Jiang L, Arya RK, Chen L, et al. Adipose Lipolysis Regulates Cardiac Glucose Uptake and Function in Mice under Cold Stress. International Journal of Molecular Sciences. 2021; 22(24):13361. https://doi.org/10.3390/ijms222413361
Chicago/Turabian StyleChoi, Youngshim, Hyunsu Shin, Ziwei Tang, Yute Yeh, Yinyan Ma, Anil K. G. Kadegowda, Huan Wang, Long Jiang, Rakesh K. Arya, Ling Chen, and et al. 2021. "Adipose Lipolysis Regulates Cardiac Glucose Uptake and Function in Mice under Cold Stress" International Journal of Molecular Sciences 22, no. 24: 13361. https://doi.org/10.3390/ijms222413361
APA StyleChoi, Y., Shin, H., Tang, Z., Yeh, Y., Ma, Y., Kadegowda, A. K. G., Wang, H., Jiang, L., Arya, R. K., Chen, L., Xue, B., Shi, H., Gavrilova, O., & Yu, L. (2021). Adipose Lipolysis Regulates Cardiac Glucose Uptake and Function in Mice under Cold Stress. International Journal of Molecular Sciences, 22(24), 13361. https://doi.org/10.3390/ijms222413361