
Systematic Review of Calcium Channels and Intracellular Calcium Signaling: Relevance to Pesticide Neurotoxicity


Abstract
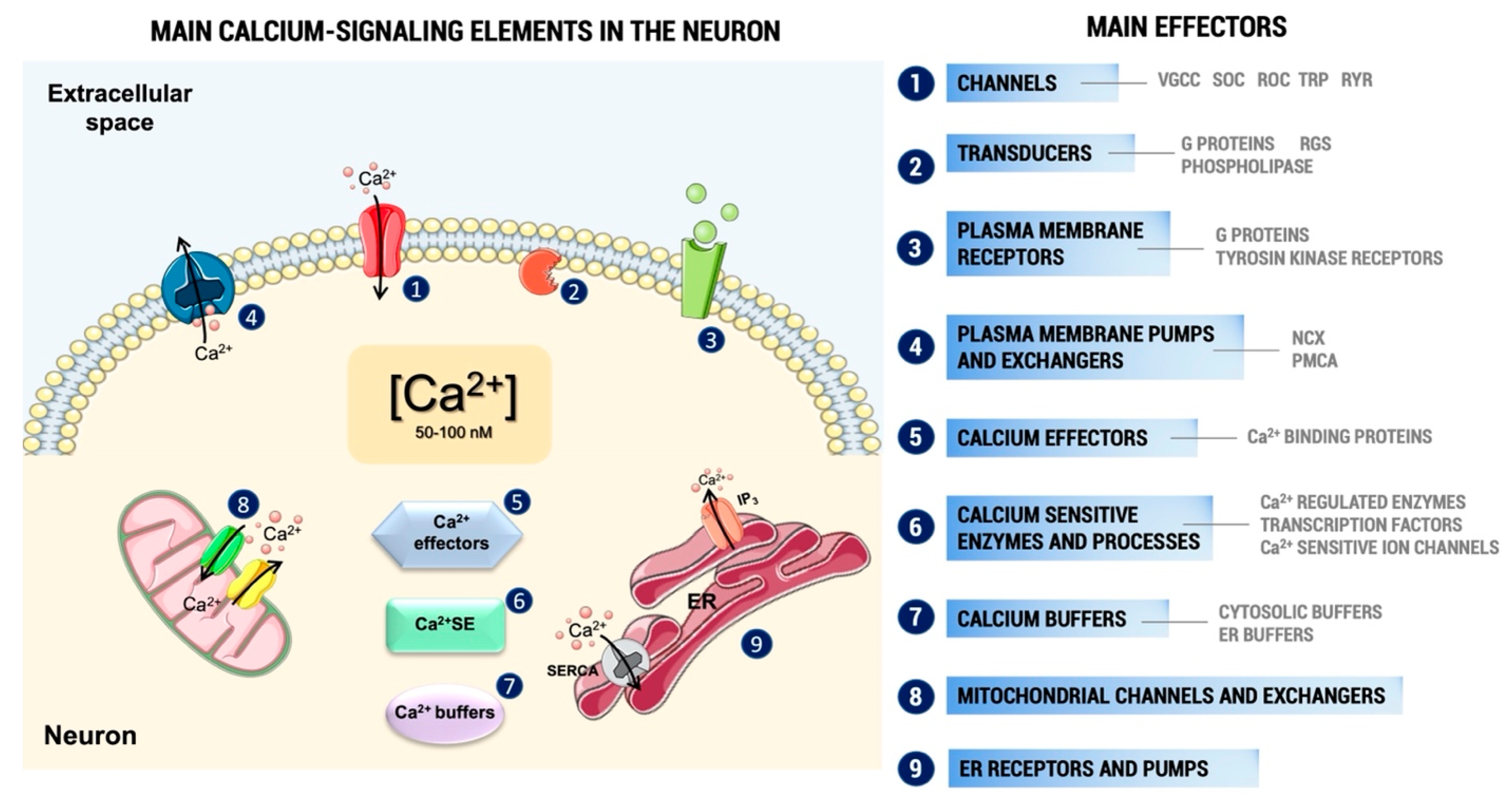
1. Introduction
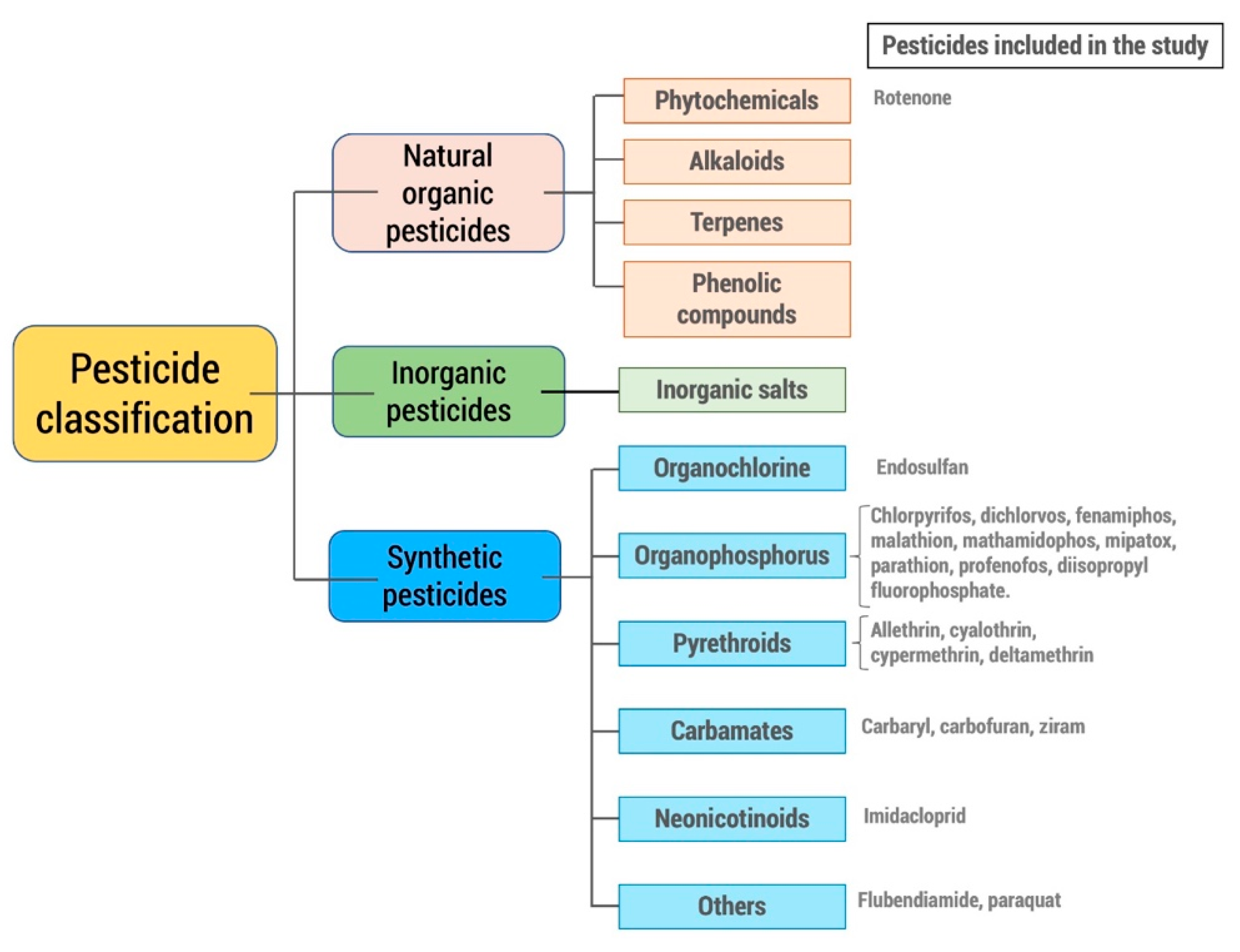
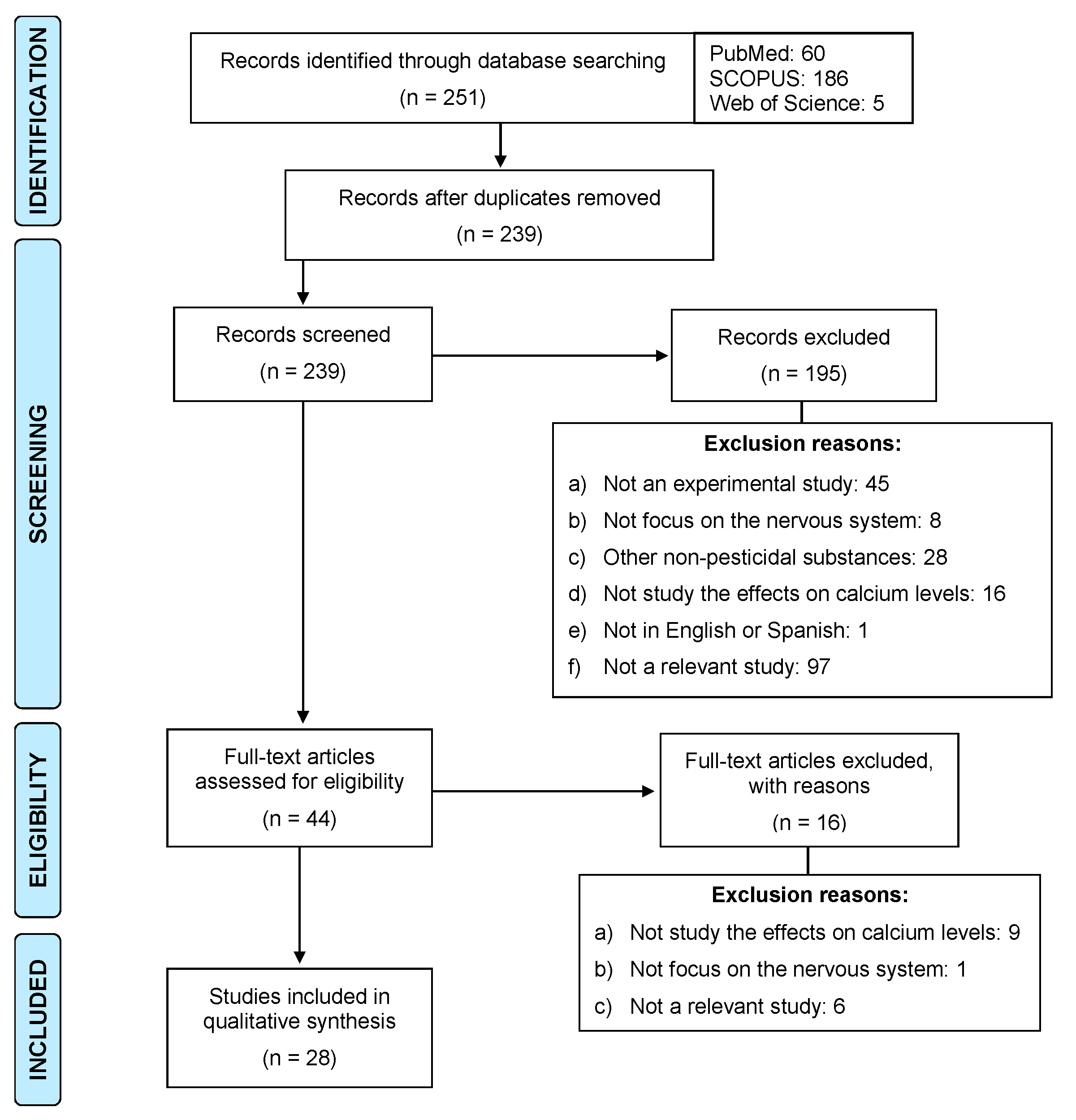
2. Methodology
Inclusion and Exclusion Criteria
3. Results
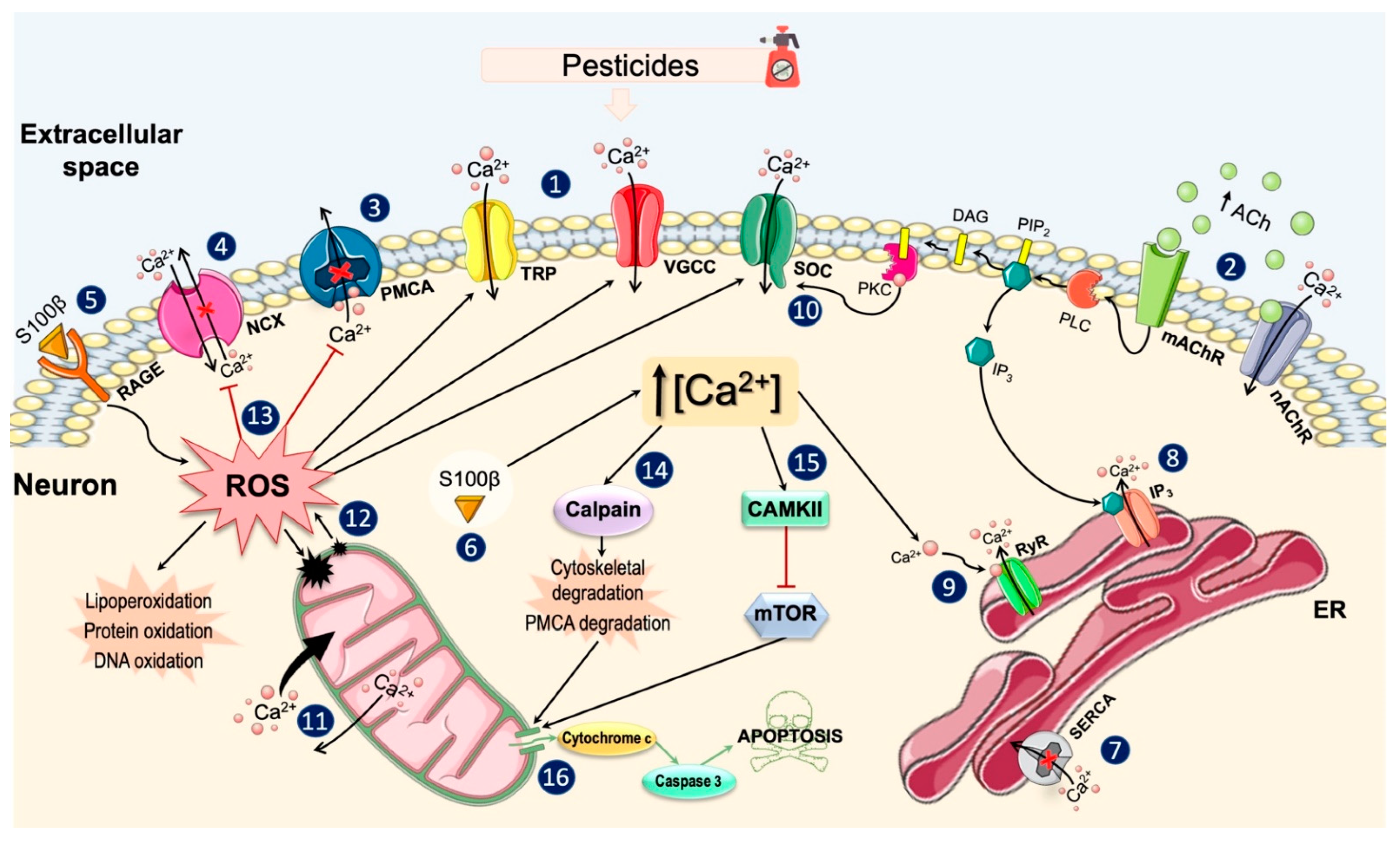
3.1. General Effects of Pesticides on Ca2+ Homeostasis
3.2. Effects on Ca2+ Channels and Plasma Membrane Ca2+ Pumps
3.3. Effects on Intracellular Ca2+ Stores
3.4. Effects on Ca2+ Binding Proteins and Intracellular Signaling Pathways
3.5. Protective Effects of Some Treatments on Pesticide-Induced Alterations in Ca2+ Homeostasis
4. Discussion
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Burgalossi, A.; Jung, S.; Man, K.-N.M.; Nair, R.; Jockusch, W.J.; Wojcik, S.M.; Brose, N.; Rhee, J.-S. Analysis of neurotransmitter release mechanisms by photolysis of caged Ca2+ in an autaptic neuron culture system. Nat. Protoc. 2012, 7, 1351–1365. [Google Scholar] [CrossRef]
- Ghosh, A.; Greenberg, M.E. Calcium signaling in neurons: Molecular mechanisms and cellular consequences. Science 1995, 268, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Engle, M.G.; Rychlik, B. Effects of elevated intracellular calcium levels on the cytoskeleton and tau in cultured human cortical neurons. Mol. Chem. Neuropathol. 1991, 15, 117–142. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Tavernarakis, N. Calcium homeostasis in aging neurons. Front. Genet. 2012, 3, 200. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Zou, M.; Lu, L.; Lau, D.; Ditzel, D.A.W.; Delucinge-Vivier, C.; Aso, Y.; Descombes, P.; Bading, H. Nuclear Calcium Signaling Controls Expression of a Large Gene Pool: Identification of a Gene Program for Acquired Neuroprotection Induced by Synaptic Activity. PLoS Genet. 2009, 5, e1000604. [Google Scholar] [CrossRef]
- Toescu, E.C. Apoptosis and cell death in neuronal cells: Where does Ca2+ fit in? Cell Calcium 1998, 24, 387–403. [Google Scholar] [CrossRef]
- Young, W. Role of calcium in central nervous system injuries. J. Neurotrauma 1992, 9 (Suppl. S1), S9–S25. [Google Scholar]
- De Coster, M.A. Calcium dynamics in the central nervous system. Adv. Neuroimmunol. 1995, 5, 233–239. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Experientia 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal Calcium Signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging Calcium in Neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef]
- El-Fawal, H.A.; Correll, L.; Gay, L.; Ehrich, M. Protease activity in brain, nerve, and muscle of hens given neuropathy-inducing organophosphates and a calcium channel blocker. Toxicol. Appl. Pharmacol. 1990, 103, 133–142. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef]
- Sandhir, R.; Gill, K.D. Alterations in calcium homeostasis on lead exposure in rat synaptosomes. Mol. Cell. Biochem. 1994, 131, 25–33. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef]
- Riascos, D.; de Leon, D.; Baker-Nigh, A.; Nicholas, A.; Yukhananov, R.; Bu, J.; Wu, C.-K.; Geula, C. Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 565–576. [Google Scholar] [CrossRef]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Baltazar, M.T.; Dinis-Oliveira, R.J.; Bastos, M.D.L.; Tsatsakis, A.; Duarte, J.A.; Carvalho, F. Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases—A mechanistic approach. Toxicol. Lett. 2014, 230, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Gilden, R.C.; Huffling, K.; Sattler, B. Pesticides and Health Risks. J. Obstet. Gynecol. Neonatal Nurs. 2010, 39, 103–110. [Google Scholar] [CrossRef]
- Mostafalou, S.; Abdollahi, M. Pesticides and human chronic diseases: Evidences, mechanisms, and perspectives. Toxicol. Appl. Pharmacol. 2013, 268, 157–177. [Google Scholar] [CrossRef]
- World Health Organization (WHO). The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification, 2019 Edition; World Health Organization: Geneva, Switzerland, 2020; Available online: https://apps.who.int/iris/handle/10665/332193 (accessed on 29 October 2021).
- Sabarwal, A.; Kumar, K.; Singh, R.P. Hazardous effects of chemical pesticides on human health–Cancer and other associated disorders. Environ. Toxicol. Pharmacol. 2018, 63, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Shah, R. Pesticides and Human Health. In Emerging Contaminants; Intechopen: London, UK, 2020. [Google Scholar] [CrossRef]
- Parrón, T.; Requena, M.; Hernández, A.F.; Alarcón, R. Association between environmental exposure to pesticides and neurodegenerative diseases. Toxicol. Appl. Pharmacol. 2011, 256, 379–385. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization; United Nations Environmental Programme. Public Health Impact of Pesticides Used in Agriculture. 1990. Available online: https://apps.who.int/iris/handle/10665/39772 (accessed on 5 December 2021).
- Von Ehrenstein, O.S.; Ling, C.; Cui, X.; Cockburn, M.; Park, A.S.; Yu, F.; Wu, J.; Ritz, B. Prenatal and infant exposure to ambient pesticides and autism spectrum disorder in children: Population based case-control study. BMJ 2019, 364, l962. [Google Scholar] [CrossRef]
- Alavanja, M.C.; Hoppin, J.A.; Kamel, F. Health Effects of Chronic Pesticide Exposure: Cancer and Neurotoxicity. Annu. Rev. Public Health 2004, 25, 155–197. [Google Scholar] [CrossRef]
- Koçyiğit, H.; Sinanoğlu, F. Method validation for the analysis of pesticide residue in aqueous environment. Environ. Monit. Assess. 2020, 192, 567. [Google Scholar] [CrossRef] [PubMed]
- Petraitis, J.; Jarmalaitė, I.; Vaičiūnas, V.; Uščinas, R.; Jankovskiene, G. A review of research studies into pesticide residues in food in Lithuania Pesticidų likučių maisto produktuose tyrimų Lietuvoje apžvalga. Zemdirbyste 2013, 100, 205–212. [Google Scholar] [CrossRef][Green Version]
- Rahman, M.; Hoque, S.; Bhowmik, S.; Ferdousi, S.; Kabiraz, M.P.; van Brakel, M.L. Monitoring of pesticide residues from fish feed, fish and vegetables in Bangladesh by GC-MS using the QuEChERS method. Heliyon 2021, 7, e06390. [Google Scholar] [CrossRef]
- Nicolopoulou-Stamati, P.; Maipas, S.; Kotampasi, C.; Stamatis, P.; Hens, L. Chemical Pesticides and Human Health: The Urgent Need for a New Concept in Agriculture. Front. Public Health 2016, 4, 148. [Google Scholar] [CrossRef]
- Bondy, S. Increased fragility of neuronal membranes with aging. Exp. Neurol. 1989, 103, 61–63. [Google Scholar] [CrossRef]
- Rosa, R.; Sanfeliu, C.; Suñol, C.; Pomés, A.; Rodríguez-Farré, E.; Schousboe, A.; Frandsen, A. The Mechanism for Hexachlorocyclohexane-Induced Cytotoxicity and Changes in Intracellular Ca2+ Homeostasis in Cultured Cerebellar Granule Neurons Is Different for the γ- and δ-Isomers. Toxicol. Appl. Pharmacol. 1997, 142, 31–39. [Google Scholar] [CrossRef]
- Younglai, E.V.; Wu, Y.; Foster, W.G. Rapid action of pesticides on cytosolic calcium concentrations in cultures of human umbilical vein endothelial cells. Reprod. Toxicol. 2006, 21, 271–279. [Google Scholar] [CrossRef]
- Wang, Q.; Diao, Q.; Dai, P.; Chu, Y.; Wu, Y.; Zhou, T.; Cai, Q. Exploring poisonous mechanism of honeybee, Apis mellifera ligustica Spinola, caused by pyrethroids. Pestic. Biochem. Physiol. 2017, 135, 1–8. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Choudhary, S.; Gill, K.D. Protective effect of nimodipine on dichlorvos-induced delayed neurotoxicity in rat brain. Biochem. Pharmacol. 2001, 62, 1265–1272. [Google Scholar] [CrossRef]
- Ding, Q.; Fang, S.; Chen, X.; Wang, Y.; Li, J.; Tian, F.; Xu, X.; Attali, B.; Xie, X.; Gao, Z. TRPA1 channel mediates organophosphate-induced delayed neuropathy. Cell Discov. 2017, 3, 17024. [Google Scholar] [CrossRef] [PubMed]
- Emerick, G.L.; Fernandes, L.S.; De Paula, E.S.; Barbosa, F., Jr.; Dos Santos, N.A.G.; Santos, A.C. In Vitro study of the neuropathic potential of the organophosphorus compounds fenamiphos and profenofos: Comparison with mipafox and paraoxon. Toxicol. Vitr. 2015, 29, 1079–1087. [Google Scholar] [CrossRef]
- Fortalezas, S.; Marques-Da-Silva, D.; Gutierrez-Merino, C. Creatine Protects Against Cytosolic Calcium Dysregulation, Mitochondrial Depolarization and Increase of Reactive Oxygen Species Production in Rotenone-Induced Cell Death of Cerebellar Granule Neurons. Neurotox. Res. 2018, 34, 717–732. [Google Scholar] [CrossRef]
- Freestone, P.S.; Chung, K.K.H.; Guatteo, E.; Mercuri, N.B.; Nicholson, L.F.; Lipski, J. Acute action of rotenone on nigral dopaminergic neurons—Involvement of reactive oxygen species and disruption of Ca2+ homeostasis. Eur. J. Neurosci. 2009, 30, 1849–1859. [Google Scholar] [CrossRef]
- Hsu, S.-S.; Jan, C.-R.; Liang, W.-Z. The investigation of the pyrethroid insecticide lambda-cyhalothrin (LCT)-affected Ca2+ homeostasis and -activated Ca2+-associated mitochondrial apoptotic pathway in normal human astrocytes: The evaluation of protective effects of BAPTA-AM (a selective Ca2+ chelator). NeuroToxicology 2018, 69, 97–107. [Google Scholar] [CrossRef]
- Hsu, S.-S.; Jan, C.-R.; Liang, W.-Z. Uncovering malathion (an organophosphate insecticide) action on Ca2+ signal transduction and investigating the effects of BAPTA-AM (a cell-permeant Ca2+ chelator) on protective responses in glial cells. Pestic. Biochem. Physiol. 2019, 157, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.S.; Hong, S.J.; Barhoumi, R.; Burghardt, R.; Donnelly, K.; Wild, J.R.; Venkatraj, V.; Tiffany-Castiglioni, E. Neurotoxicity induced in differentiated SK-N-SH-SY5Y human neuroblastoma cells by organophosphorus compounds. Toxicol. Appl. Pharmacol. 2003, 186, 110–118. [Google Scholar] [CrossRef]
- Kamboj, A.; Sandhir, R. Perturbed Synaptosomal Calcium Homeostasis and Behavioral Deficits Following Carbofuran Exposure: Neuroprotection by N-Acetylcysteine. Neurochem. Res. 2007, 32, 507–516. [Google Scholar] [CrossRef]
- Li, H.Y.; Zhong, Y.F.; Wu, S.Y.; Shi, N. NF-E2 Related Factor 2 Activation and Heme Oxygenase-1 Induction by tert-Butylhydroquinone Protect against Deltamethrin-Mediated Oxidative Stress in PC12 Cells. Chem. Res. Toxicol. 2007, 20, 1242–1251. [Google Scholar] [CrossRef]
- Liu, C.; Ye, Y.; Zhou, Q.; Zhang, R.; Zhang, H.; Liu, W.; Xu, C.; Liu, L.; Huang, S.; Chen, L. Crosstalk between Ca2+ signaling and mitochondrial H2O2 is required for rotenone inhibition of mTOR signaling pathway leading to neuronal apoptosis. Oncotarget 2016, 7, 7534–7549. [Google Scholar] [CrossRef] [PubMed]
- Raheja, G.; Gill, K.D. Calcium homeostasis and dichlorvos induced neurotoxicity in rat brain. Mol. Cell. Biochem. 2002, 232, 13–18. [Google Scholar] [CrossRef]
- Bondy, S.C.; Halsall, L. Lindane-induced modulation of calcium levels within synaptosomes. NeuroToxicology 1988, 9, 645–652. [Google Scholar]
- Pomés, A.; Frandsen, A.A.; Suñol, C.; Sanfeliu, C.; Rodriguez-Farre, E.; Schousboe, A. Lindane cytotoxicity in cultured neocortical neurons is ameliorated by GABA and flunitrazepam. J. Neurosci. Res. 1994, 39, 663–668. [Google Scholar] [CrossRef]
- Sun, X.; Liu, X.-B.; Martinez, J.; Zhang, G.H. Effects of low concentrations of paraoxon on Ca2+ mobilization in a human parotid salivary cell-line HSY. Arch. Oral Biol. 2000, 45, 621–638. [Google Scholar] [CrossRef]
- Meijer, M.; Hamers, T.; Westerink, R.H. Acute disturbance of calcium homeostasis in PC12 cells as a novel mechanism of action for (sub)micromolar concentrations of organophosphate insecticides. NeuroToxicology 2014, 43, 110–116. [Google Scholar] [CrossRef]
- Qian, Y.; Venkatraj, J.; Barhoumi, R.; Pal, R.; Datta, A.; Wild, J.R.; Tiffany-Castiglioni, E. Comparative non-cholinergic neurotoxic effects of paraoxon and diisopropyl fluorophosphate (DFP) on human neuroblastoma and astrocytoma cell lines. Toxicol. Appl. Pharmacol. 2007, 219, 162–171. [Google Scholar] [CrossRef]
- Kadala, A.; Charreton, M.; Collet, C. Flubendiamide, the first phthalic acid diamide insecticide, impairs neuronal calcium signalling in the honey bee’s antennae. J. Insect Physiol. 2020, 125, 104086. [Google Scholar] [CrossRef]
- Martin, C.A.; Myers, K.M.; Chen, A.; Martin, N.T.; Barajas, A.; Schweizer, F.E.; Krantz, D.E. Ziram, a pesticide associated with increased risk for Parkinson’s disease, differentially affects the presynaptic function of aminergic and glutamatergic nerve terminals at the Drosophila neuromuscular junction. Exp. Neurol. 2015, 275, 232–241. [Google Scholar] [CrossRef]
- Vatanparast, J.; Janahmadi, M.; Asgari, A.R. Involvement of protein kinase C and IP3-mediated Ca2+ release in activity modulation by paraoxon in snail neurons. Eur. J. Pharmacol. 2007, 571, 81–87. [Google Scholar] [CrossRef]
- Vatanparast, J.; Janahmadi, M.; Asgari, A.R.; Sepehri, H.; Haeri-Rohani, A. Paraoxon suppresses Ca2+ spike and afterhyperpolarization in snail neurons: Relevance to the hyperexcitability induction. Brain Res. 2006, 1083, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Suzuki, T.; Unno, T.; Komori, S.; Kobayashi, H. Differential presynaptic actions of pyrethroid insecticides on glutamatergic and GABAergic neurons in the hippocampus. Toxicology 2008, 243, 155–163. [Google Scholar] [CrossRef]
- Jin, J.; Lao, A.; Katsura, M.; Caputo, A.; Schweizer, F.; Sokolow, S. Involvement of the sodium–calcium exchanger 3 (NCX3) in ziram-induced calcium dysregulation and toxicity. NeuroToxicology 2014, 45, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Meijer, M.; Brandsema, J.A.R.; Nieuwenhuis, D.; Wijnolts, F.M.J.; Dingemans, M.M.L.; Westerink, R.H.S. Inhibition of voltage-gated calcium channels after subchronic and repeated exposure of PC12 cells to different classes of insecticides. Toxicol. Sci. 2015, 147, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.-K.; Kim, I.; Zhao, P.; Estacion, M.; Black, J.A.; Waxman, S.G. Sodium Channels Contribute to Degeneration of Dorsal Root Ganglion Neurites Induced by Mitochondrial Dysfunction in an In Vitro Model of Axonal Injury. J. Neurosci. 2013, 33, 19250–19261. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Fernandes, D.; Bean, J.L.; Michaelis, M.L. Effects of paraquat-induced oxidative stress on the neuronal plasma membrane Ca2+-ATPase. Free Radic. Biol. Med. 2009, 47, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Emerick, G.L.; Ehrich, M.; Jortner, B.S.; Oliveira, R.; DeOliveira, G.H. Biochemical, histopathological and clinical evaluation of delayed effects caused by methamidophos isoforms and TOCP in hens: Ameliorative effects using control of calcium homeostasis. Toxicology 2012, 302, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Dolphin, A.C. Functions of Presynaptic Voltage-gated Calcium Channels. Function 2020, 2, zqaa027. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Verma, S.K.; Raheja, G.; Kaur, P.; Joshi, K.; Gill, K.D. The L-Type Calcium Channel Blocker Nimodipine Mitigates Cytoskeletal Proteins Phosphorylation in Dichlorvos-Induced Delayed Neurotoxicity in Rats. Basic Clin. Pharmacol. Toxicol. 2006, 98, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, J.V. Differences in Sensitivity of Children and Adults to Chemical Toxicity: The NAS Panel Report. Regul. Toxicol. Pharmacol. 2000, 31, 280–285. [Google Scholar] [CrossRef]
- Heusinkveld, H.J.; Westerink, R.H.S. Organochlorine Insecticides Lindane and Dieldrin and Their Binary Mixture Disturb Calcium Homeostasis in Dopaminergic PC12 Cells. Environ. Sci. Technol. 2012, 46, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Heusinkveld, H.J.; Molendijk, J.; Berg, M.V.D.; Westerink, R.H.S. Azole Fungicides Disturb Intracellular Ca2+ in an Additive Manner in Dopaminergic PC12 Cells. Toxicol. Sci. 2013, 134, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Faber, E.S.L.; Sah, P. Calcium-Activated Potassium Channels: Multiple Contributions to Neuronal Function. Neuroscientist 2003, 9, 181–194. [Google Scholar] [CrossRef]
- Lipscombe, D.; Allen, S.E.; Toro, C. Control of neuronal voltage-gated calcium ion channels from RNA to protein. Trends Neurosci. 2013, 36, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Heppner, T.J.; Fiekers, J.F. Soman reversibly decreases the duration of Ca2+ and Ba2+ action potentials in bullfrog sympathetic neurons. Brain Res. 1991, 563, 303–305. [Google Scholar] [CrossRef]
- Heppner, T.J.; Fiekers, J.F. Vx enhances neuronal excitability and alters membrane properties of Rana catesbeiana sympathetic ganglion neurons. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1992, 102, 335–338. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Store-Operated Calcium Channels: New Perspectives on Mechanism and Function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003970. [Google Scholar] [CrossRef]
- Dhaka, A.; Viswanath, V.; Patapoutian, A. Trp ion channels and temperature sensation. Annu. Rev. Neurosci. 2006, 29, 135–161. [Google Scholar] [CrossRef]
- Yassaka, R.T.; Inagaki, H.; Fujino, T.; Nakatani, K.; Kubo, T. Enhanced activation of the transient receptor potential channel TRPA1 by ajoene, an allicin derivative. Neurosci. Res. 2010, 66, 99–105. [Google Scholar] [CrossRef]
- Belrose, J.C.; Jackson, M.F. TRPM2: A candidate therapeutic target for treating neurological diseases. Acta Pharmacol. Sin. 2018, 39, 722–732. [Google Scholar] [CrossRef]
- Faouzi, M.; Penner, R. TRPM2. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Springer: Berlin/Heidelberg, Germany, 2014; pp. 403–426. [Google Scholar] [CrossRef]
- Costa, L.G. Current issues in organophosphate toxicology. Clin. Chim. Acta 2006, 366, 1–13. [Google Scholar] [CrossRef]
- Abdallah, E.A.M.; Jett, D.A.; Eldefrawi, M.E.; Eldefrawi, A.T. Differential effects of paraoxon on the M3 muscarinic receptor and its effector system in rat submaxillary gland cells. J. Biochem. Toxicol. 1992, 7, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bakry, N.M.S.; El-Rashidy, A.H.; Eldefrawi, A.T.; Eldefrawi, M.E. Direct actions of organophosphate anticholinesterases on nicotinic and muscarinic acetylcholine receptors. J. Biochem. Toxicol. 1988, 3, 235–259. [Google Scholar] [CrossRef]
- Jett, D.; Abdallah, E.; El-Fakahany, E.; Eldefrawi, M.; Eldefrawi, A. High-affinity activation by paraoxon of a muscarinic receptor subtype in rat brain striatum. Pestic. Biochem. Physiol. 1991, 39, 149–157. [Google Scholar] [CrossRef]
- Katz, L.S.; Marquis, J.K. Organophosphate-induced alterations in muscarinic receptor binding and phosphoinositide hydrolysis in the human SK-N-SH cell line. NeuroToxicology 1992, 13, 365–378. [Google Scholar] [PubMed]
- Oortgiesen, M.; Van Kleef, R.G.; Zwart, R.; Beukel, I.V.D.; Vijverberg, H.P. Nicotinic Receptors of Different Species Exhibit Differential Sensitivities to Nitromethylene and Organophosphate Insecticides. Altern. Lab. Anim. 1996, 24, 367–376. [Google Scholar] [CrossRef]
- Beukel, I.V.D.; Van Kleef, R.G.; Oortgiesen, M. Direct Functional Effects of Parathion and Paraoxon on Neuronal Nicotinic and Muscarinic M3 Acetylcholine Receptors. Altern. Lab. Anim. 1996, 24, 191–199. [Google Scholar] [CrossRef]
- Viana, G.; Davis, L.; Kauffman, F. Effects of organophosphates and nerve growth factor on muscarinic receptor binding number in rat pheochromocytoma PC12 cells. Toxicol. Appl. Pharmacol. 1988, 93, 257–266. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Iacobucci, G.J.; Popescu, G.K. NMDA receptors: Linking physiological output to biophysical operation. Nat. Rev. Neurosci. 2017, 18, 236–249. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Díaz Horta, O. El ion calcio: Su regulación y función en la célula ß pancreática. Rev. Cuba. Endocrinol. 2003, 14. [Google Scholar]
- Brini, M.; Carafoli, E. Calcium Pumps in Health and Disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Brini, M.; Carafoli, E. The PMCA pumps in genetically determined neuronal pathologies. Neurosci. Lett. 2018, 663, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. Intracellular calcium homeostasis. Annu. Rev. Biochem. 1987, 56, 395–433. [Google Scholar] [CrossRef]
- Shenoda, B. The Role of Na+/Ca2+ Exchanger Subtypes in Neuronal Ischemic Injury. Transl. Stroke Res. 2015, 6, 181–190. [Google Scholar] [CrossRef]
- Jeffs, G.J.; Meloni, B.P.; Bakker, A.J.; Knuckey, N.W. The role of the Na+/Ca2+ exchanger (NCX) in neurons following ischaemia. J. Clin. Neurosci. 2007, 14, 507–514. [Google Scholar] [CrossRef]
- Lytton, J. Na+/Ca2+ Exchangers and Ca2+ Transport in Neurons. In Handbook of Neurochemistry and Molecular Neurobiology; Lajtha, A., Reith, M.E.A., Eds.; Springer: Boston, MA, USA, 2007; pp. 225–241. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Deniaud, A.; El Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C.A. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2007, 27, 285–299. [Google Scholar] [CrossRef]
- Verkhratsky, A. Endoplasmic reticulum calcium signaling in nerve cells. Biol. Res. 2004, 37, 693–699. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Lisbona, F.; Rivera, D.R.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1833, 3507–3517. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Kaur, P.; Radotra, B.; Minz, R.; Gill, K.D. Impaired mitochondrial energy metabolism and neuronal apoptotic cell death after chronic dichlorvos (OP) exposure in rat brain. NeuroToxicology 2007, 28, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.D.; Falcão, K.V.; Assis, C.R.; Martins, R.M.; Araújo, M.C.; Yogui, G.T.; Neves, J.L.; Seabra, G.M.; Maia, M.B.; Amaral, I.P.; et al. Effects of pyriproxyfen on zebrafish brain mitochondria and acetylcholinesterase. Chemosphere 2020, 263, 128029. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2017, 70, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Murphy, A.N.; Bredesen, D.E.; Cortopassi, G.; Wang, E.; Fiskum, G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. USA 1996, 93, 9893–9898. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Tan, J.; Wan, Z.; Zou, Y.; Afewerky, H.K.; Zhang, Z.; Zhang, T. Effects of Commonly Used Pesticides in China on the Mitochondria and Ubiquitin-Proteasome System in Parkinson’s Disease. Int. J. Mol. Sci. 2017, 18, 2507. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Mehrpour, O.; Forouzanfar, F.; Roshanravan, B.; Samarghandian, S. Oxidative stress and mitochondrial dysfunction in organophosphate pesticide-induced neurotoxicity and its amelioration: A review. Environ. Sci. Pollut. Res. 2020, 27, 24799–24814. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.C.; Meyer, J. Mitochondria as a target of organophosphate and carbamate pesticides: Revisiting common mechanisms of action with new approach methodologies. Reprod. Toxicol. 2019, 89, 83–92. [Google Scholar] [CrossRef]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Pickett, C.; Li, S.; Jones, J.; Chen, H.; Webb, B.; Choi, J.; Zhou, Y.; Zimmerman, M.C.; et al. Compartmentalized oxidative stress in dopaminergic cell death induced by pesticides and complex I inhibitors: Distinct roles of superoxide anion and superoxide dismutases. Free Radic. Biol. Med. 2013, 61, 370–383. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W.; Tomita, T. Phospholipase C signaling and calcium influx. Adv. Biol. Regul. 2012, 52, 152–164. [Google Scholar] [CrossRef]
- Bodjarian, N.; Carpentier, P.; Blanchet, G.; Baubichon, D.; Lallement, G. Cholinergic activation of phosphoinositide metabolism during soman-induced seizures. Neuroreport 1993, 4, 1191–1193. [Google Scholar]
- Bodjarian, N.; Carpentier, P.; Baubichon, D.; Blanchet, G.; Lallement, G. Involvement of non-muscarinic receptors in phosphoinositide signalling during soman-induced seizures. Eur. J. Pharmacol. Mol. Pharmacol. 1995, 289, 291–297. [Google Scholar] [CrossRef]
- Mobley, P. The cholinesterase inhibitor soman increases inositol trisphosphate in rat brain. Neuropharmacology 1990, 29, 189–191. [Google Scholar] [CrossRef]
- Patro, N.; Shrivastava, M.; Tripathi, S.; Patro, I.K. S100β upregulation: A possible mechanism of deltamethrin toxicity and motor coordination deficits. Neurotoxicol. Teratol. 2009, 31, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H.; Taylor, P. Muscarinic receptor agonists and antagonists. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 9th ed.; Hardman, J.G., Limbird, L.E., Eds.; McGraw-Hill: New York, NY, USA, 1996; pp. 141–160. [Google Scholar]
- Gresset, A.; Sondek, J.; Harden, T.K. The Phospholipase C Isozymes and Their Regulation. In Phosphoinositides I: Enzymes of Synthesis and Degradation; Springer: Dordrecht, The Netherlands, 2012; Volume 58, pp. 61–94. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Putney, J. Differential Effects of Protein Kinase C Activation on Calcium Storage and Capacitative Calcium Entry in NIH 3T3 Cells. J. Biol. Chem. 1996, 271, 21522–21528. [Google Scholar] [CrossRef] [PubMed]
- Lipp, P.; Reither, G. Protein Kinase C: The “Masters” of Calcium and Lipid. Cold Spring Harb. Perspect. Biol. 2011, 3, a004556. [Google Scholar] [CrossRef]
- Cullen, P.J. Calcium Signalling: The Ups and Downs of Protein Kinase C. Curr. Biol. 2003, 13, R699–R701. [Google Scholar] [CrossRef]
- Mellor, H.; Parker, P. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [CrossRef]
- Carragher, N.O. Calpain Inhibition: A Therapeutic Strategy Targeting Multiple Disease States. Curr. Pharm. Des. 2006, 12, 615–638. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A. Calpain Dysregulation in Alzheimer’s Disease. ISRN Biochem. 2012, 2012, 728571. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Hayward, N.J.; Elliott, P.J.; Sawyer, S.D.; Baker, K.L.; Dean, R.L.; Akiyama, A.; Straub, J.A.; Harbeson, S.L.; Li, Z. Calpain inhibitor AK295 protects neurons from focal brain ischemia. Effects of postocclusion intra-arterial administration. Stroke 1994, 25, 2265–2270. [Google Scholar] [CrossRef]
- Potz, B.A.; Abid, M.R.; Sellke, F.W. Role of Calpain in Pathogenesis of Human Disease Processes. J. Nat. Sci. 2016, 2, e218. [Google Scholar] [PubMed]
- Yamashima, T. Ca2+-dependent proteases in ischemic neuronal death: A conserved ‘calpain-cathepsin cascade’ from nematodes to primates. Cell Calcium 2004, 36, 285–293. [Google Scholar] [CrossRef]
- Abou-Donia, M.B. The cytoskeleton as a target for organophosphorus ester-induced delayed neurotoxicity (OPIDN). Chem. Interact. 1993, 87, 383–393. [Google Scholar] [CrossRef]
- Jortner, B.S. Mechanisms of Toxic Injury in the Peripheral Nervous System: Neuropathologic Considerations. Toxicol. Pathol. 2000, 28, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, W.W.; Hasler, M.B. Characterization of the calcium-induced disruption of neurofilaments in rat peripheral nerve. Brain Res. 1979, 168, 299–309. [Google Scholar] [CrossRef]
- Wang, K.K.; Villalobo, A.; Roufogalis, B.D. Activation of the Ca2+-ATPase of human erythrocyte membrane by an endogenous Ca2+-dependent neutral protease. Arch. Biochem. Biophys. 1988, 260, 696–704. [Google Scholar] [CrossRef]
- Lushington, G.H.; Zaidi, A.; Michaelis, M.L. Theoretically predicted structures of plasma membrane Ca2+-ATPase and their susceptibilities to oxidation. J. Mol. Graph. Model. 2005, 24, 175–185. [Google Scholar] [CrossRef]
- Zaidi, A.; Barŕon, L.; Sharov, V.S.; Schöneich, C.; Michaelis, E.K.; Michaelis, M.L. Oxidative Inactivation of Purified Plasma Membrane Ca2+-ATPase by Hydrogen Peroxide and Protection by Calmodulin. Biochemistry 2003, 42, 12001–12010. [Google Scholar] [CrossRef] [PubMed]
- Junho, C.V.C.; Caio-Silva, W.; Trentin-Sonoda, M.; Carneiro-Ramos, M.S. An Overview of the Role of Calcium/Calmodulin-Dependent Protein Kinase in Cardiorenal Syndrome. Front. Physiol. 2020, 11, 735. [Google Scholar] [CrossRef]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Zalcman, G.; Federman, N.; Romano, A. CaMKII Isoforms in Learning and Memory: Localization and Function. Front. Mol. Neurosci. 2018, 11, 445. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Qin, J.; Ma, J.; Zeng, Q.; Zhang, H.; Zhang, R.; Liu, C.; Xu, C.; Zhang, S.; Huang, S.; et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca2+-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell. Signal. 2018, 53, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Rothermundt, M.; Peters, M.; Prehn, J.; Arolt, V. S100B in brain damage and neurodegeneration. Microsc. Res. Tech. 2003, 60, 614–632. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W. S100 proteins structure functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar] [CrossRef]
- Thelin, E.P.; Nelson, D.W.; Bellander, B.-M. A review of the clinical utility of serum S100B protein levels in the assessment of traumatic brain injury. Acta Neurochir. 2016, 159, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1793, 1008–1022. [Google Scholar] [CrossRef]
- Michetti, F.; Corvino, V.; Geloso, M.C.; Lattanzi, W.; Bernardini, C.; Serpero, L.; Gazzolo, D. The S100B protein in biological fluids: More than a lifelong biomarker of brain distress. J. Neurochem. 2012, 120, 644–659. [Google Scholar] [CrossRef] [PubMed]
- Hajduková, L.; Sobek, O.; Prchalová, D.; Bilkova, Z.; Koudelková, M.; Lukášková, J.; Matuchová, I. Biomarkers of Brain Damage: S100B and NSE Concentrations in Cerebrospinal Fluid—A Normative Study. BioMed Res. Int. 2015, 2015, 379071. [Google Scholar] [CrossRef] [PubMed]
- Michetti, F.; D’Ambrosi, N.; Toesca, A.; Puglisi, M.A.; Serrano, A.; Marchese, E.; Corvino, V.; Geloso, M.C. The S100B story: From biomarker to active factor in neural injury. J. Neurochem. 2018, 148, 168–187. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.; Van Eldik, L. S100 beta stimulates calcium fluxes in glial and neuronal cells. J. Biol. Chem. 1992, 267, 9689–9694. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Abdollahi, M.; Ranjbar, A.; Shadnia, S.; Nikfar, S.; Rezaie, A. Pesticides and oxidative stress: A review. Med. Sci. Monit. 2004, 10, RA141–RA147. [Google Scholar]
- Corcoran, G.; Wong, B.K.; Neese, B.L. Early sustained rise in total liver calcium during acetaminophen hepatotoxicity in mice. Res. Commun. Chem. Pathol. Pharmacol. 1987, 58, 291–305. [Google Scholar]
- Sevillano, S.; De La Mano, A.; De Dios, I.; Ramudo, L.; Manso, M. Major pathological mechanisms of acute pancreatitis are prevented by N-acetylcysteine. Digestion 2003, 68, 34–40. [Google Scholar] [CrossRef]
- Alamed, J.; Chaiyasit, W.; McClements, D.; Decker, E.A. Relationships between Free Radical Scavenging and Antioxidant Activity in Foods. J. Agric. Food Chem. 2009, 57, 2969–2976. [Google Scholar] [CrossRef]
- Shih, A.Y.; Li, P.; Murphy, T. A Small-Molecule-Inducible Nrf2-Mediated Antioxidant Response Provides Effective Prophylaxis against Cerebral Ischemia In Vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef]
- Wang, Z.; Ji, C.; Wu, L.; Qiu, J.; Li, Q.; Shao, Z.; Chen, G. Tert-Butylhydroquinone Alleviates Early Brain Injury and Cognitive Dysfunction after Experimental Subarachnoid Hemorrhage: Role of Keap1/Nrf2/ARE Pathway. PLoS ONE 2014, 9, e97685. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Zhao, W.; Liu, M.; Liu, L.; Wang, Y. TBHQ-Overview of Multiple Mechanisms against Oxidative Stress for Attenuating Methamphetamine-Induced Neurotoxicity. Oxidative Med. Cell. Longev. 2020, 2020, 8874304. [Google Scholar] [CrossRef]
- Granatiero, V.; Konrad, C.; Bredvik, K.; Manfredi, G.; Kawamata, H. Nrf2 signaling links ER oxidative protein folding and calcium homeostasis in health and disease. Life Sci. Alliance 2019, 2, e201900563. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci. Rep. 2019, 9, 10366. [Google Scholar] [CrossRef]
- Roschel, H.; Gualano, B.; Ostojic, S.M.; Rawson, E.S. Creatine Supplementation and Brain Health. Nutrients 2021, 13, 586. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.N.; Agharkar, A.S.; Gonzales, E.B. A review of creatine supplementation in age-related diseases: More than a supplement for athletes. F1000Research 2014, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.D.; Bröer, S. Creatine as a booster for human brain function. How might it work? Neurochem. Int. 2015, 89, 249–259. [Google Scholar] [CrossRef]
- Dean, P.J.A.; Arikan, G.; Opitz, B.; Sterr, A. Potential for use of creatine supplementation following mild traumatic brain injury. Concussion 2017, 2, CNC34. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, C.; Néant, I.; Moreau, M. The calcium: An early signal that initiates the formation of the nervous system during embryogenesis. Front. Mol. Neurosci. 2012, 5, 3. [Google Scholar] [CrossRef]
- Lohmann, C. Calcium signaling and the development of specific neuronal connections. Prog. Brain Res. 2009, 175, 443–452. [Google Scholar] [CrossRef]
- León-Iza, M.; Zarain-Herzberg, Á. El ion calcio como segundo mensajero en el desarrollo del sistema nervioso. Rev. Med. UV 2011, 11, 32–39. [Google Scholar]
- Verkhratsky, A.J.; Petersen, O.H. Neuronal calcium stores. Cell Calcium 1998, 24, 333–343. [Google Scholar] [CrossRef]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmström, K.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The Ins and Outs of Mitochondrial Calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.-I.; Jou, M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Iza, S.C.C.; Rodríguez, A.I. Oxidative stress and pesticide disease: A challenge for toxicology. Rev. Fac. Med. 2018, 66, 261–267. [Google Scholar] [CrossRef]
- Ledda, C.; Cannizzaro, E.; Cinà, D.; Filetti, V.; Vitale, E.; Paravizzini, G.; Di Naso, C.; Iavicoli, I.; Rapisarda, V. Oxidative stress and DNA damage in agricultural workers after exposure to pesticides. J. Occup. Med. Toxicol. 2021, 16, 1. [Google Scholar] [CrossRef]
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium 2016, 60, 108–114. [Google Scholar] [CrossRef]
- Momeni, H.R. Role of Calpain in Apoptosis. Cell J. 2011, 13, 65–72. [Google Scholar]
- Abiria, S.A.; Colbran, R.J. CaMKII associates with CaV1.2 L-type calcium channels via selected β subunits to enhance regulatory phosphorylation. J. Neurochem. 2009, 112, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; Abiria, S.A.; Wu, Y.; Anderson, M.E.; Colbran, R.J. Differential Regulated Interactions of Calcium/Calmodulin-Dependent Protein Kinase II with Isoforms of Voltage-Gated Calcium Channel β Subunits. Biochemistry 2008, 47, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.S.; Lim, I.A.; Hemsworth, D.E.; Horne, M.; Hell, J.W. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 3239–3244. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species or Cellular Line | Dose and Time of Exposure | Objective | Results | Reference |
|---|---|---|---|---|
| Wistar rat | Dichlorvos: 6 mg/kg s.c. for 8 weeks | To investigate alterations in neuronal Ca2+ homeostasis | ↑ [Ca2+]i in brain stem and cerebellum | [50] |
| Wistar rat | Dichlorvos: 200 mg/kg s.c. in a single dose | To examine the role of the Ca2+ messenger system in the development of delayed neurotoxicity | ↑ [Ca2+]i | [39] |
| Wistar rat | Carbofuran: 1 mg/kg oral for 28 days | To study the alterations in Ca2+ homeostasis and neurobehavioral deficits induced by the pesticide | ↑ [Ca2+]i in the synaptosomes | [47] |
| Rat CGN | Rotenone: 2–50 nM for 30 min or 12 h | To assess the effects of rotenone at very low concentrations in mature CGN | ↑ [Ca2+]i induced by rotenone (10 and 15 nM) at 30 min | [42] |
| Rat midbrain slices | Rotenone: 0.05–1 μM for 10 min | Investigate the effects of rotenone on individual neurons of the rat substantia nigra pars compacta | ↑ [Ca2+]i | [43] |
| Mouse DRG | Malathion: 0.1–100 μM for 0–16 min | To assess the role of TRPA1 in organophosphate-induced delayed neuropathy | ↑ [Ca2+]i and upregulation of neuronal excitability | [40] |
| Mouse PC12 cells and primary neurons | Rotenone: 0–1 μM for 24 h | To investigate whether rotenone induces apoptosis by inhibition of the Ca2+/ROS-dependent mTOR pathway | ↑ [Ca2+]i ↑mitochondrial H2O2 levels, which induced ↑ [Ca2+]i | [49] |
| PC12 cells | Carbaryl, chlorpyrifos, parathion-ethyl and its metabolites–oxon: 0.1–10 µM for 20 min | To evaluate the effects of several pesticides and their metabolites on the basal [Ca2+]i | - All OPs inhibited the depolarization-induced ↑ [Ca2+]i - The parent compounds were more potent than their –oxon metabolites in altering the [Ca2+]i - The mixtures of chlorpyrifos+oxon analog or +parathion did not increase the degree of inhibition on the ↑ [Ca2+]i | [54] |
| PC12 cells | DLT: 10 µM for 1 h | To investigate the neuroprotective effect of tert-butylhydroquinone against oxidative stress induced by DLT | ↑ [Ca2+]i | [48] |
| GHA and human glioblastoma DBTRG-05MG cells, and D1 TNC1 rat astrocytes | Malathion: 5–25 µM | To explore the mechanism underlying the effects of malathion on Ca2+ homeostasis and cell viability | Concentration-dependent ↑ [Ca2+]i in GHA cells | [45] |
| GHA and D1 TNC1 cells | LCT: 10–15 µM | To explore whether LCT affects Ca2+ homeostasis and cell viability | ↑ [Ca2+]i | [44] |
| SH-SY5Y | Mipaxon, paraoxon: 0.05–2 mM for 4 days | To characterize the cellular targets of organophosphate neurotoxicity | - Paraoxon induced a transient ↑ [Ca2+]i - Repeated treatment with paraoxon (0.05 mM) ↓ [Ca2+]i in the NGF-differentiated cells | [46] |
| SH-SY5Y | Mipafox, paraoxon, fenamiphos, profenofos: 1 × 10−10–1 × 10−2 M for 24 or 48 h | To evaluate the neurotoxic effects of mipafox and paraoxon, as well as the potential of fenamiphos and profenofos to cause acute and/or delayed effects | Both mipafox and fenamiphos ↑ [Ca2+]i | [41] |
| SH-SY5Y and CCF-STTG1 | Paraoxon, DFP: 0.3, 1, 3, 10 or 30 μM for 1–4 days | To compare the neurotoxic effects of paraoxon and DFP in two cell lines | Paraoxon (1–30 µM), but not DFP, ↓ the mitochondrial:cytosolic Ca2+ ratio in TLC cultures | [55] |
| Domestic honeybees (Apis mellifera) | Flubendiamide: 3 µM | To evaluate the effects of the insecticide on normal Ca2+ homeostasis in antennal neurons of honeybees | Strong Ca2+ transients in antennal neurons | [56] |
| Apis mellifera ligustica Spinola | DLT: 0–250 mg/L for 200 seg or 5 min | To investigate the effect of DLT on the Ca2+ channel in nerve cells of the brain | ↑ [Ca2+]i even with the lowest pesticide concentrations | [37] |
| Drosophila melanogaster | Ziram: 20 μM | To compare the effects of ziram on type II aminergic versus type Ib glutamatergic nerve endings | Spontaneous and synchronized bursts of Ca2+ input and electrical activity in type II, but not in type Ib terminals | [57] |
| Snail neurons | Paraoxon: 0.3–0.6 µM for 10 min | To investigate the interaction of paraoxon with PKC and the release of Ca2+ mediated by IP3, on the modulation of action potentials and neuronal activity | ↓ Duration of Ca2+ action potentials and ↓ duration of PHP, associated with an ↑ in firing frequency Paraoxon (0.6 μM) ↓ the duration of action potentials, but ↑ the duration of PHP, along with a ↓ in the firing rate | [58] |
| Neuronal soma of land snail (Caucasotachea atrolabiata) | Paraoxon: 0.3 µM for 5 or 10 min | To study the effects of the pesticide on Ca2+ spikes and neuronal excitability in snail neurons | Paraoxon (0.3 μM) reversibly ↓ the duration and amplitude of the Ca2+ peaks ↓ in the duration and amplitude of PHP, leading to a significant ↑ in the frequency of Ca2+ peaks | [59] |
| Species or Cellular Line | Dose and Time of Exposure | Results | Reference |
|---|---|---|---|
| Wistar rat | Dichlorvos: 6 mg/kg s.c. for 8 weeks | ↑ Ca2+ influx through the VGCC ↓ Ca2+-ATPase activity | [50] |
| Wistar rat | Dichlorvos: 200 mg/kg s.c. in a single dose | ↓ Ca2+-ATPase activity | [39] |
| Sprague Dawley rat | Allethrin, cyhalothrin, DLT: 10, 20 or 60 mg/kg i.p. in a single dose | Nimodipine completely blocked the glutamate release induced by DLT (60 mg/kg) | [60] |
| Wistar rat | Carbofuran: 1 mg/kg oral for 28 days | ↓ Ca2+-ATPase activity with a concomitant ↑ in K+-induced Ca2+ influx through VGCCs | [47] |
| Mouse primary ventral midbrain neurons | Ziram: 10 mM | Dopaminergic neurons lacking NCX3 were less sensitive to ziram-induced neurotoxicity | [61] |
| PC12 cells | Carbaryl, chlorpyrifos, parathion-ethyl and its metabolites –oxon: 0.1–10 μM for 20 min | - The parent compounds were more potent than their –oxon metabolites in altering the [Ca2+]i - The mixtures of chlorpyrifos+oxon analog or +parathion did not increase the degree of inhibition on the ↑ [Ca2+]i | [54] |
| PC12 cells and rat primary cortical cells | Endosulfan, cypermethrin, chlorpyrifos, chlorpyrifos-oxon, carbaryl, and IMI: 0.1–100 μM for 24 h (and 20 min in the second exposure) | - All insecticides (except carbaryl and IMI) induced slow or non-reversible VGCCs inhibition (subchronic conditions) - Chlorpyrifos was clearly more potent in inhibiting VGCCs in the repeated exposure compared to acute exposure | [62] |
| Mouse DRG | Rotenone: 1 μM for 3 or 6 days | - NCX reverse mode inhibition protected against rotenone-exposed neurites from degeneration - Rotenone exposure was associated with delayed Ca2+ elimination after neurite activation | [63] |
| Rat CGN cells | Rotenone: 2–50 nM for 30 min or 12 h | Nifedipine and, to a lesser extent, MK-801 attenuated the rotenone-induced alteration in the Ca2+ homeostasis | [42] |
| Rat midbrain slices | Rotenone: 0.05–1 μM for 10 min | The rotenone-induced ↑ [Ca2+]i was blocked by eliminating extracellular Ca2+ and was attenuated by a TRPM2 blocker | [43] |
| Rat primary cortical neurons | Paraquat: 5–100 μM for 5 min, 15 min or 24 h | - Paraquat (5–10 μM) doubled the basal activity of PMCA, but abolished its sensitivity to calmodulin - Paraquat (25–100 μM) ↓ PMCA activity and were associated with the formation of high molecular weight PMCA aggregates | [64] |
| Mouse DRG | Malathion: 0.1–100 μM for 0–16 min | Malathion-induced Ca2+ influx currents were attenuated by a TRPA1 antagonist and eliminated by suppression of Trpa1.gene | [40] |
| Hens (Leghorn isabrown) | Methamidophos: 50 mg/kg oral for 1 or 21 days | Nimodipine ↓ alterations induced by the pesticide | [65] |
| GHA and human glioblastoma DBTRG-05MG cells, and D1 TNC1 rat astrocytes | Malathion: 5–25 μM | - The malathion-induced ↑ [Ca2+]i was reduced by eliminating the Ca2+ from extracellular medium - Malathion-induced ↑ [Ca2+]i was inhibited by blockers of SOCs | [45] |
| GHA and D1 TNC1 cells | LCT: 10–15 μM | LCT-induced ↑ [Ca2+]i was reduced by eliminating extracellular Ca2+ and was inhibited by modulators of SOCs | [44] |
| SH-SY5Y | Mipaxon, Paraoxon: 0.05–2 mM for 4 days | Paraoxon (0.05 mM) attenuated the transient ↑ [Ca2+]i induced by carbachol | [46] |
| Apis mellifera ligustica Spinola | DLT: 0–250 mg/L for 200 seg or 5 min | DLT had toxic effects on T-type VGCCs, but not on L-type VGCCs, channels activated by NMDAR or Ca2+ store | [37] |
| Neuronal soma of land snail (Caucasotachea atrolabiata) | Paraoxon: 0.3 μM for 5 or 10 min | - Apamine ↓ the duration and amplitude of PHP and ↑ the frequency of the peaks - In the presence of apamine, paraoxon ↓ the duration of the Ca2+ peak and PHP and ↑ the frequency of neuronal activation | [59] |
| Species or Cellular Line | Dose and Time of Exposure | Results | Reference |
|---|---|---|---|
| Wistar rat | Dichlorvos: 6 mg/kg s.c. for 12 weeks | ↑ influx of Ca2+ to mitochondria | [109] |
| GHA and human glioblastoma DBTRG-05MG cells, and D1 TNC1 rat astrocytes | Malathion: 5–25 μM | In a Ca2+-free medium, the pretreatment with tapsigargin, a SERCA inhibitor, abolished the pesticide-induced ↑ [Ca2+]i Incubation with malathion abolished the tapsigargin-induced ↑ [Ca2+]i | [45] |
| GHA and D1 TNC1 cells | LCT: 10–15 μM | In a Ca2+-free medium the pretreatment with tapsigargin suppressed LCT-induced ↑ [Ca2+]i Incubation with LCT abolished the tapsigargin-induced ↑ [Ca2+]i | [44] |
| Zebrafish (Danio rerio) | Pyriproxyfen: 0.001–10 μmol/L for 1 h (in vitro essay) 0.001, 0.01 or 0.1 μg/mL for 16 h (in vivo essay) | Pyriproxyfen (0.1 μg/mL) ↓ Ca2+ uptake by up to 50% Pyridoxyphene (0.01 or 0.1 μg/mL) ↓ mitochondrial Ca2+ release by approximately 80% | [110] |
| Species or Cellular Line | Dose and Time of Exposure | Results | Reference |
|---|---|---|---|
| Wistar rat | Dichlorvos: 6 mg/kg s.c. for 8 weeks | ↑ calpain activity | [50] |
| Wistar rat | Dichlorvos: 200 mg/kg s.c. Single dose | ↑ calpain activity | [39] |
| Wistar rat | DLT: 0.7 mg/kg i.p. from PND0 until PND7 (DLT-I) or from PND9 until PND13 (DLT-II) | DLT ↑ expression of S100β in radial glial fibers and in astrocytes on PND12 and PND15 days The up-regulation of S100β was more prominent in the DLT-II group | [125] |
| Rat primary cortical neurons | Paraquat: 5–100 μM for 5 min, 15 min or 24 h | Proteolytic degradation of PMCA was prevented by a calpain inhibitor | [64] |
| PC12 cells and rat primary cortical cells | Rotenone: 0–1 μM for 24 h | Rotenone-induced ↑ [Ca2+]i activated CaMKII and caused inhibition of mTOR signaling | [49] |
| Hens (Leghorn isabrown) | Methamidophos: 50 mg/kg oral for 1 or 21 days | The (+) and (-) isoforms of methamidophos produced a slight ↑ in calpain activity | [65] |
| GHA and human glioblastoma DBTRG-05MG cells, and D1 TNC1 rat astrocytes | Malathion: 5–25 μM | Inhibition of PLC blocked the ↑ [Ca2+]i induced by malathion In Ca2+ medium, malathion-induced ↑ [Ca2+]i was inhibited by a PKC inhibitor | [45] |
| SH-SY5Y | Mipafox, paraoxon, fenamiphos, profhenophs: 1 × 10−10–1 × 10−2 M for 24 or 48 h | Mipafox induced calpain activation after 24 h | [41] |
| Snail neurons | Paraoxon: 0.3–0.6 μM for 10 min | - Modulation of PKC activity modified Ca2+ action potentials and neuronal activity, but did not contribute to the neurotoxic actions of paraoxon - TMB-8, an IP3 receptor-mediated intracellular Ca2+ release antagonist, suppressed the paraoxon-induced secondary increase in the duration of PHP and neuronal silencing | [58] |
| Species or Cellular Line | Dose and Time of Exposure | Results | Reference |
|---|---|---|---|
| Wistar rat | Carbofuran: 1 mg/kg oral for 28 days | NAC had a beneficial effect on Ca2+ homeostasis | [47] |
| Rat CGN cells | Rotenone: 2–50 nM for 30 min or 12 h | Creatine attenuated early rotenone-induced [Ca2+]i dysregulation | [42] |
| PC12 cells | DLT: 10 μM for 1 h | Tert-butylhydroquinone reduced the ↑ [Ca2+]i induced by DLT | [48] |
| PC12 cells and rat primary cortical cells | Rotenone: 0–1 μM for 24 h | Chelation of the [Ca2+]i with BAPTA-AM or prevention of extracellular Ca2+ entry by EGTA ↓ H2O2 overproduction | [49] |
| GHA and human glioblastoma DBTRG-05MG cells, and D1 TNC1 rat astrocytes | Malathion: 5–25 μM | Chelation of cytosolic Ca2+ with BAPTA-AM prevented the malathion-induced cytotoxicity | [45] |
| GHA and D1 TNC1 cells | LCT: 10–15 μM | Chelation of cytosolic Ca2+ with BAPTA-AM ↓ the apoptosis induced by LCT | [44] |
| Neuronal soma of land snail (Caucasotachea atrolabiata) | Paraoxon: 0.3 μM for 5 or 10 min | BAPTA-AM ↓ the duration and amplitude of PHP and ↑ the duration and frequency of Ca2+ peaks In the presence of BAPTA-AM, paraoxon ↓ the duration of Ca2+ peaks without affecting their frequency | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costas-Ferreira, C.; Faro, L.R.F. Systematic Review of Calcium Channels and Intracellular Calcium Signaling: Relevance to Pesticide Neurotoxicity. Int. J. Mol. Sci. 2021, 22, 13376. https://doi.org/10.3390/ijms222413376
Costas-Ferreira C, Faro LRF. Systematic Review of Calcium Channels and Intracellular Calcium Signaling: Relevance to Pesticide Neurotoxicity. International Journal of Molecular Sciences. 2021; 22(24):13376. https://doi.org/10.3390/ijms222413376
Chicago/Turabian StyleCostas-Ferreira, Carmen, and Lilian R. F. Faro. 2021. "Systematic Review of Calcium Channels and Intracellular Calcium Signaling: Relevance to Pesticide Neurotoxicity" International Journal of Molecular Sciences 22, no. 24: 13376. https://doi.org/10.3390/ijms222413376
APA StyleCostas-Ferreira, C., & Faro, L. R. F. (2021). Systematic Review of Calcium Channels and Intracellular Calcium Signaling: Relevance to Pesticide Neurotoxicity. International Journal of Molecular Sciences, 22(24), 13376. https://doi.org/10.3390/ijms222413376