
Primary Amine Nucleophilic Addition to Nitrilium Closo-Dodecaborate [B12H11NCCH3]−: A Simple and Effective Route to the New BNCT Drug Design

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
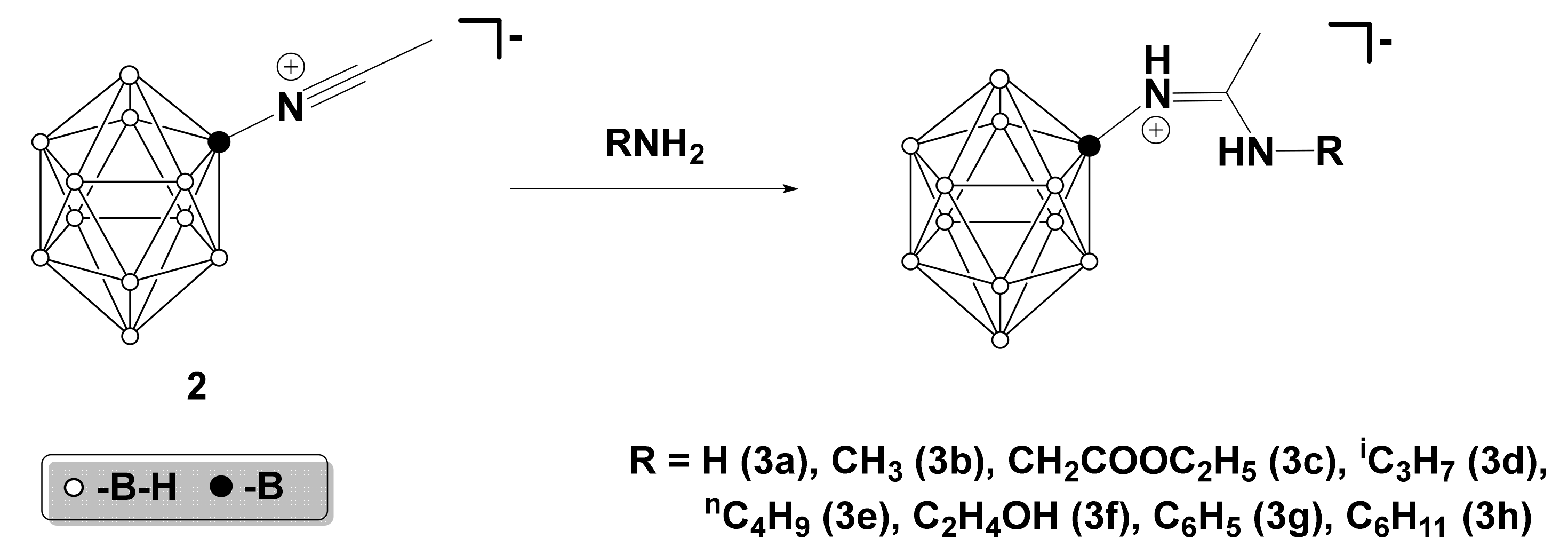
2.1. Synthesis of Nitrilium Derivatives of the Closo-Dodecaborate Anion
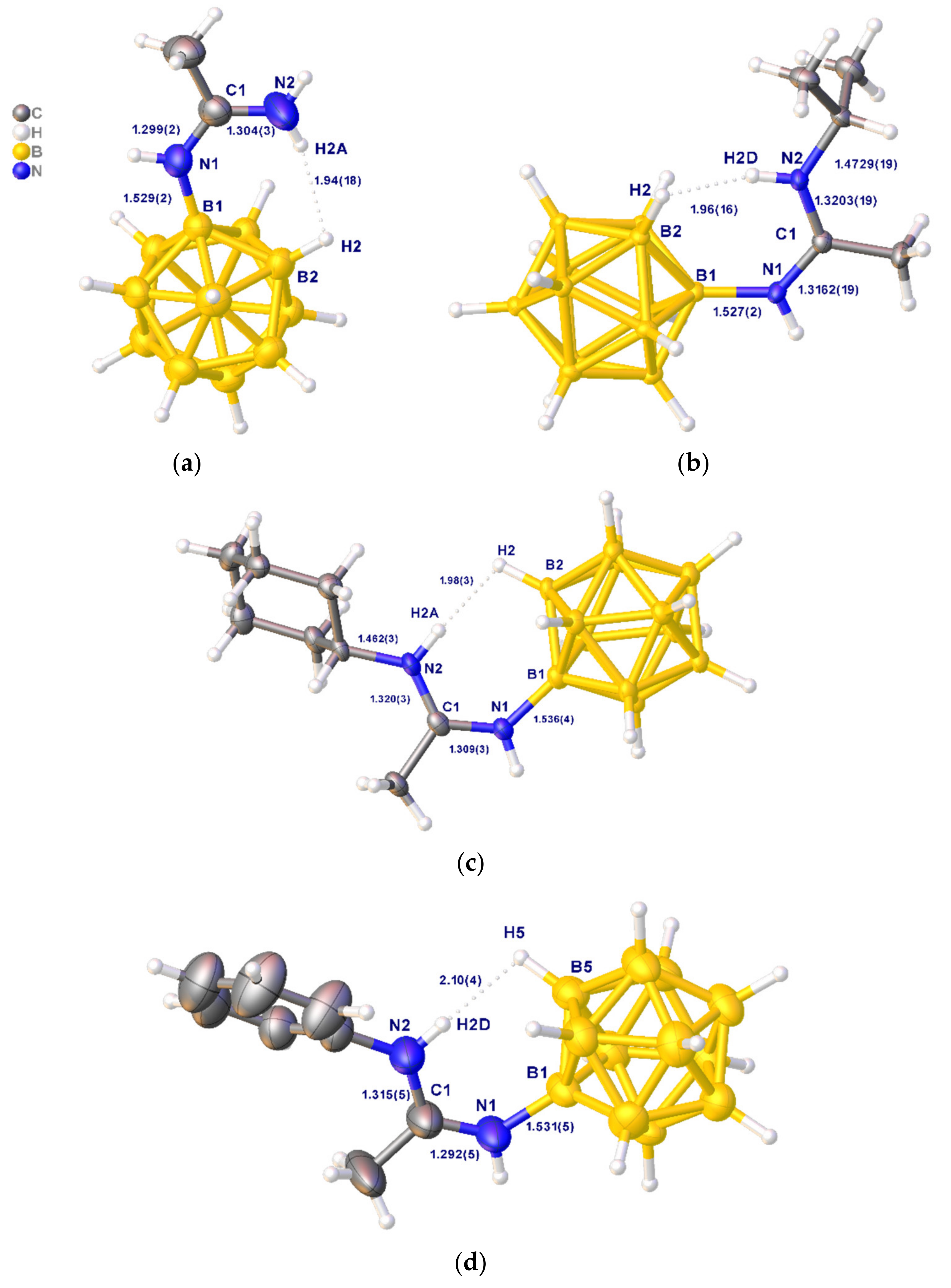
2.2. Interaction with Primary Amines
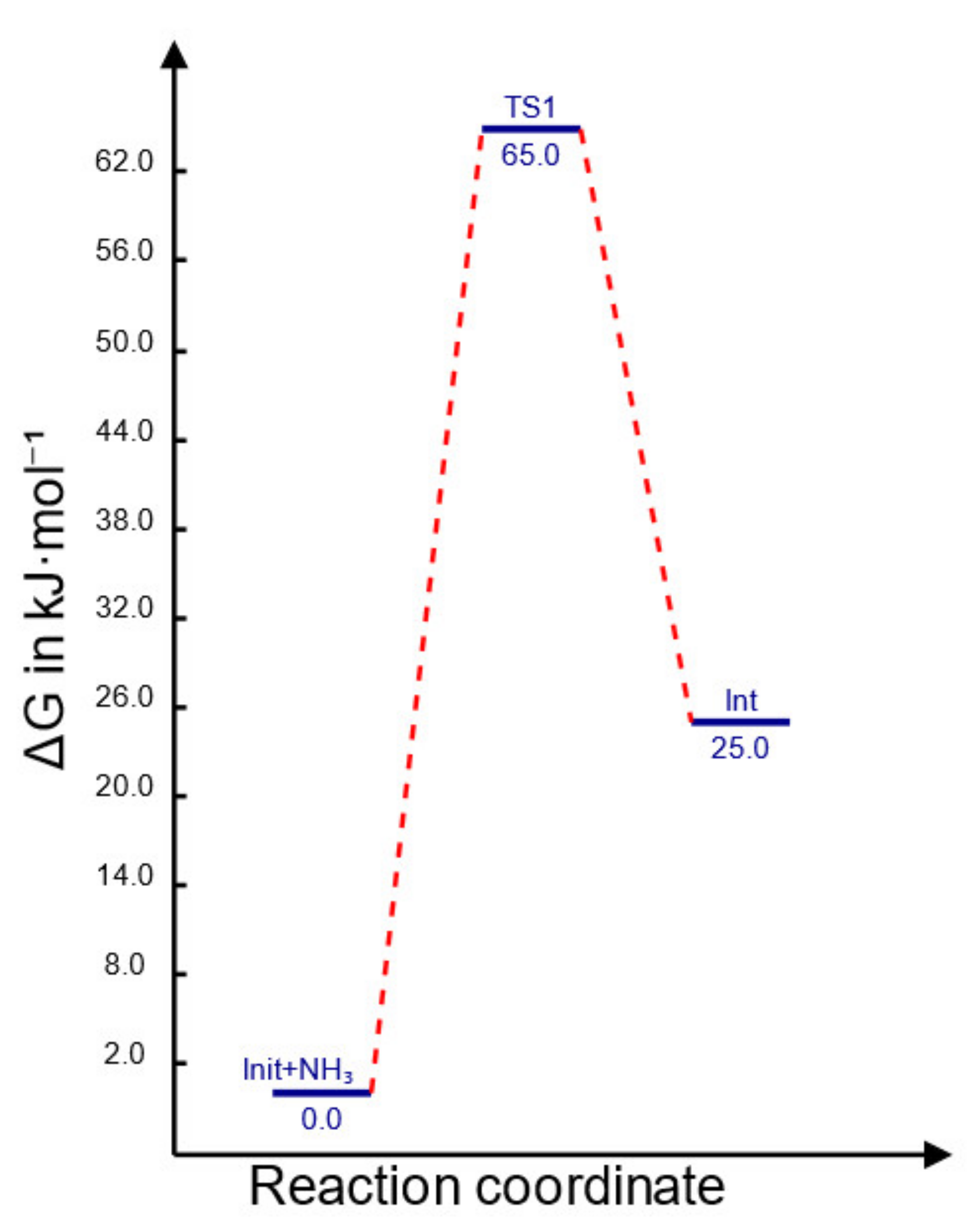
2.3. Theoretical Investigation of Addition Process Mechanism
2.4. Biological Activity Study
2.4.1. MTT Assay
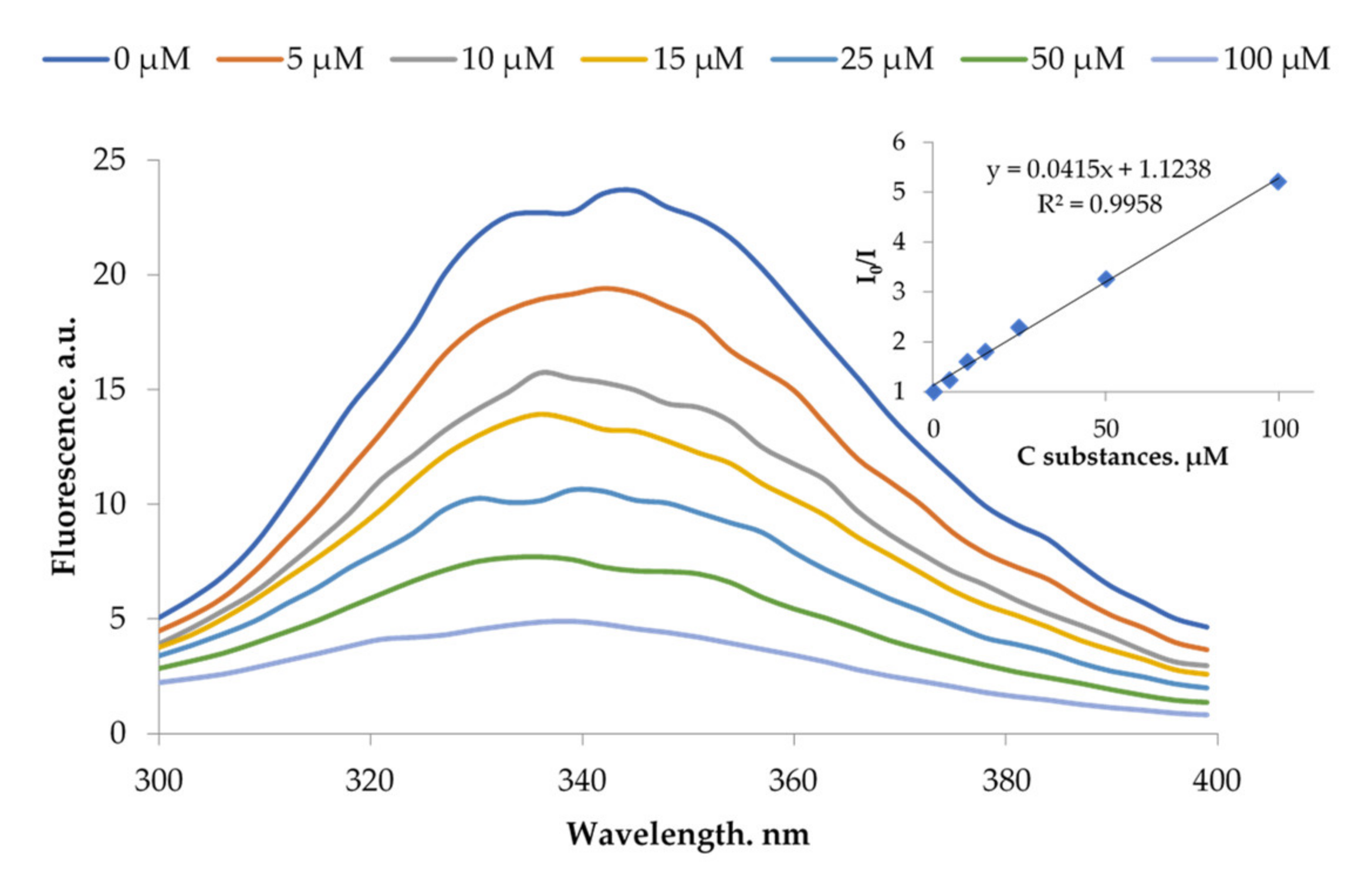
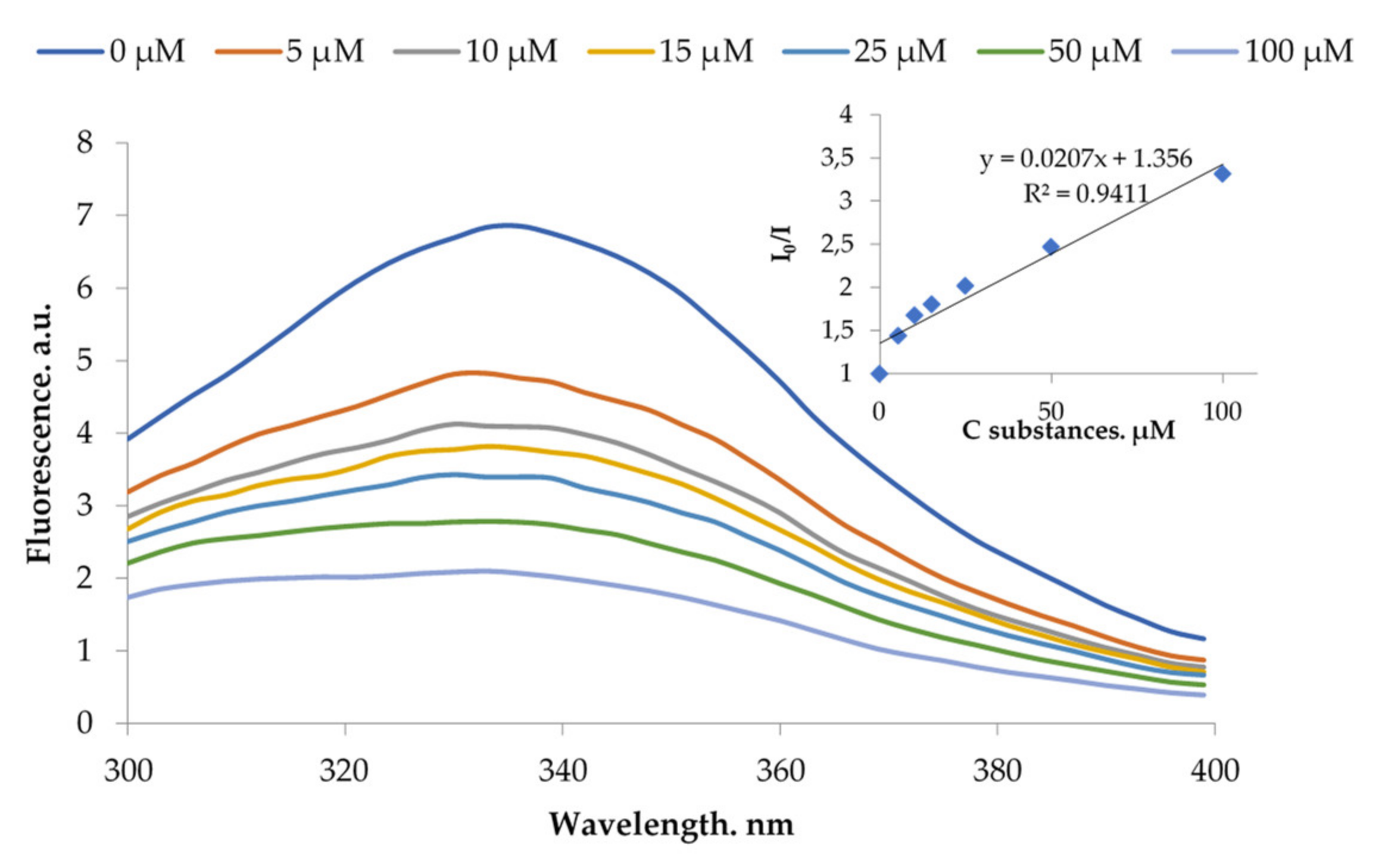
2.4.2. Complex Formation with BSA and HSA
3. Materials and Methods
3.1. Synthesis of (NBu4)[B12H11(NCCH3)]
3.2. Synthesis of Sodium Salts for Biological Assays
3.3. Biological Study
3.3.1. Cell Cultures
3.3.2. MTT Assay
3.3.3. Fluorescence Quenching Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, G.; Zhang, Y.; Wang, J. Recent Advances in the Synthesis of Aryl Nitrile Compounds. Adv. Synth. Catal. 2017, 359, 4068–4105. [Google Scholar] [CrossRef]
- Sugai, T.; Yamazaki, T.; Yokoyama, M.; Ohta, H. Biocatalysis in Organic Synthesis: The Use of Nitrile- and Amide-hydrolyzing Microorganisms. Biosci. Biotechnol. Biochem. 1997, 61, 1419–1427. [Google Scholar] [CrossRef]
- Chen, J.; Song, Q.; Wang, C.; Xi, Z. Novel cycloaddition of nitriles with monolithio- and dilithiobutadienes. J. Am. Chem. Soc. 2002, 124, 6238–6239. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qi, C.; Wang, L.; Guo, Y.; Li, D.; Jiang, H. Base-Promoted Three-Component Cascade Reaction of α-Hydroxy Ketones, Malonodinitrile, and Alcohols: Direct Access to Tetrasubstituted NH-Pyrroles. J. Org. Chem. 2021, 86, 9610–9620. [Google Scholar] [CrossRef] [PubMed]
- Sruthi, P.R.; Anas, S. An overview of synthetic modification of nitrile group in polymers and applications. J. Polym. Sci. 2020, 58, 1039–1061. [Google Scholar] [CrossRef]
- Guchhait, S.K.; Sisodiya, S.; Saini, M.; Shah, Y.V.; Kumar, G.; Daniel, D.P.; Hura, N.; Chaudhary, V. Synthesis of Polyfunctionalized Pyrroles via a Tandem Reaction of Michael Addition and Intramolecular Cyanide-Mediated Nitrile-to-Nitrile Condensation. J. Org. Chem. 2018, 83, 5807–5815. [Google Scholar] [CrossRef] [PubMed]
- Pollard, V.A.; Fuentes, M.-A.; Robertson, S.D.; Weetman, C.; Kennedy, A.R.; Brownlie, J.; Angus, F.J.; Smylie, C.; Mulvey, R.E. Reactivity studies and structural outcomes of a bulky dialkylaluminium amide in the presence of the N-heterocyclic carbene, ItBu. Polyhedron 2021, 209, 115469. [Google Scholar] [CrossRef]
- Van Dijk, T.; Chris Slootweg, J.; Lammertsma, K. Nitrilium ions-synthesis and applications. Org. Biomol. Chem. 2017, 15, 10134–10144. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Goffitzer, D.J.; Xiang, M.; Chen, Y.; Jiang, W.; Diefenbach, M.; Zhu, H.; Holthausen, M.C.; Roesky, H.W. 1-Aza-2,4-disilabicyclo[1.1.0]butanes with Superelongated C–N σ-Bonds. J. Am. Chem. Soc. 2021, 143, 8244–8248. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.E.; Hammond, C.A.; Oberle, K.G.; Ramey, E.E.; Zou, Y.; Lash, R.C.; Turlington, C.R. Nitrile Oxidation at a Ruthenium Complex leading to Intermolecular Imido Group Transfer. Organometallics 2020, 39, 3775–3779. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-F.; Yang, T.; Tsang, L.-Y.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Azavinylidene Complexes from Coupling Reactions of Organonitriles with Phosphines. Organometallics 2021, 40, 358–369. [Google Scholar] [CrossRef]
- Garduño, J.A.; García, J.J. Toward Amines, Imines, and Imidazoles: A Viewpoint on the 3d Transition-Metal-Catalyzed Homogeneous Hydrogenation of Nitriles. ACS Catal. 2020, 10, 8012–8022. [Google Scholar] [CrossRef]
- Guo, B.; de Vries, J.G.; Otten, E. Hydration of nitriles using a metal–ligand cooperative ruthenium pincer catalyst. Chem. Sci. 2019, 10, 10647–10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himo, F.; Demko, Z.P.; Noodleman, L.; Sharpless, K.B. Why is tetrazole formation by addition of azide to organic nitriles catalyzed by zinc(II) salts? J. Am. Chem. Soc. 2003, 125, 9983–9987. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Nerush, A.; Iron, M.A.; Leitus, G.; Diskin-Posner, Y.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. Activation of nitriles by metal ligand cooperation. reversible formation of ketimido- and enamido-rhenium PNP pincer complexes and relevance to catalytic design. J. Am. Chem. Soc. 2013, 135, 17004–17018. [Google Scholar] [CrossRef] [PubMed]
- Bokach, N.A.; Kukushkin, V.Y. Addition of HO-nucleophiles to free and coordinated nitriles. Russ. Chem. Rev. 2005, 74, 153–170. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Additions to metal-activated organonitriles. Chem. Rev. 2002, 102, 1771–1802. [Google Scholar] [CrossRef] [PubMed]
- Michelin, R.A.; Mozzon, M.; Bertani, R. Reactions of transition metal-coordinated nitriles. Coord. Chem. Rev. 1996, 147, 299–338. [Google Scholar] [CrossRef]
- Chin, C.P.; Ren, Y.; Berry, J.; Knott, S.A.; McLauchlan, C.C.; Szczepura, L.F. Small molecule activation of nitriles coordinated to the [Re6Se8]2+ core: Formation of oxazine, oxazoline and carboxamide complexes. Dalton Trans. 2018, 47, 4653–4660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, W.; Freund, C.; Santos, A.M.; Herdtweck, E.; Mink, J.; Kühn, F.E. Cationic copper (I) and silver (I) nitrile complexes with fluorinated weakly coordinating anions: Metal–nitrile bond strength and its influence on the catalytic performance. Inorg. Chim. Acta 2006, 359, 4723–4729. [Google Scholar] [CrossRef]
- Díez, Á.; Forniés, J.; Fuertes, S.; Lalinde, E.; Larraz, C.; López, J.A.; Martín, A.; Moreno, M.T.; Sicilia, V. Synthesis and luminescence of cyclometalated compounds with nitrile and isocyanide ligandst. Organometallics 2009, 28, 1705–1718. [Google Scholar] [CrossRef]
- Malinowski, J.; Zych, D.; Jacewicz, D.; Gawdzik, B.; Drzeżdżon, J. Application of coordination compounds with transition metal ions in the chemical industry—A review. Int. J. Mol. Sci. 2020, 21, 5443. [Google Scholar] [CrossRef] [PubMed]
- Wilding, B.; Veselá, A.B.; Perry, J.J.B.; Black, G.W.; Zhang, M.; Martínková, L.; Klempier, N. An investigation of nitrile transforming enzymes in the chemo-enzymatic synthesis of the taxol sidechain. Org. Biomol. Chem. 2015, 13, 7803–7812. [Google Scholar] [CrossRef] [Green Version]
- Stogniy, M.Y.; Erokhina, S.A.; Sivaev, I.B.; Bregadze, V.I. Nitrilium derivatives of polyhedral boron compounds (boranes, carboranes, metallocarboranes): Synthesis and reactivity. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 983–988. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Anisimov, A.A.; Sivaev, I.B.; Bregadze, V.I. Nucleophilic addition reactions to the ethylnitrilium derivative of: Nido -carborane 10-EtCN-7,8-C2B9H11. New J. Chem. 2018, 42, 17958–17967. [Google Scholar] [CrossRef]
- El Anwar, S.; Růžičková, Z.; Bavol, D.; Fojt, L.; Grüner, B. Tetrazole Ring Substitution at Carbon and Boron Sites of the Cobalt Bis(dicarbollide) Ion Available via Dipolar Cycloadditions. Inorg. Chem. 2020, 59, 17430–17442. [Google Scholar] [CrossRef]
- Černý, R.; Brighi, M.; Murgia, F. The Crystal Chemistry of Inorganic Hydroborates. Chemistry 2020, 2, 805–826. [Google Scholar] [CrossRef]
- Nakagawa, F.; Kawashima, H.; Morita, T.; Nakamura, H. Water-Soluble closo-Docecaborate-Containing Pteroyl Derivatives Targeting Folate Receptor-Positive Tumors for Boron Neutron Capture Therapy. Cells 2020, 9, 1615. [Google Scholar] [CrossRef]
- Fisher, S.P.; Tomich, A.W.; Lovera, S.O.; Kleinsasser, J.F.; Guo, J.; Asay, M.J.; Nelson, H.M.; Lavallo, V. Nonclassical Applications of closo-Carborane Anions: From Main Group Chemistry and Catalysis to Energy Storage. Chem. Rev. 2019, 119, 8262–8290. [Google Scholar] [CrossRef]
- Keener, M.; Hunt, C.; Carroll, T.G.; Kampel, V.; Dobrovetsky, R.; Hayton, T.W.; Ménard, G. Redox-switchable carboranes for uranium capture and release. Nature 2020, 577, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Alamón, C.; Dávila, B.; García, M.F.; Sánchez, C.; Kovacs, M.; Trias, E.; Barbeito, L.; Gabay, M.; Zeineh, N.; Gavish, M.; et al. Sunitinib-Containing Carborane Pharmacophore with the Ability to Inhibit Tyrosine Kinases Receptors FLT3, KIT and PDGFR-β, Exhibits Powerful In Vivo Anti-Glioblastoma Activity. Cancers 2020, 12, 3423. [Google Scholar] [CrossRef] [PubMed]
- Bakardjiev, M.; El Anwar, S.; Bavol, D.; Růžičková, Z.; Grűner, B. Focus on Chemistry of the 10-Dioxane-nido-7,8-dicarba-undecahydrido Undecaborate Zwitterion; Exceptionally Easy Abstraction of Hydrogen Bridge and Double-Action Pathways Observed in Ring Cleavage Reactions with OH− as Nucleophile. Molecules 2020, 25, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, F.; Hosmane, N.S.; Zhu, Y. Boron Chemistry for Medical Applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, Y.; Zhang, H.; Zhou, X. Cubic platinum nanoparticles capped with Cs2[closo-B12H12] as an effective oxidation catalyst for converting methane to ethanol. J. Colloid Interface Sci. 2020, 566, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Ma, X.; Liu, Y.; Zhang, H. Direct conversion of methane into methanol and ethanol via spherical Au@Cs2[closo-B12H12] and Pd@Cs2[closo-B12H12] nanoparticles. Int. J. Hydrogen Energy 2021, 46, 30750–30761. [Google Scholar] [CrossRef]
- Brighi, M.; Murgia, F.; Łodziana, Z.; Schouwink, P.; Wołczyk, A.; Černý, R. A mixed anion hydroborate/carba-hydroborate as a room temperature Na-ion solid electrolyte. J. Power Sources 2018, 404, 7–12. [Google Scholar] [CrossRef]
- Duchêne, L.; Remhof, A.; Hagemann, H.; Battaglia, C. Status and prospects of hydroborate electrolytes for all-solid-state batteries. Energy Storage Mater. 2020, 25, 782–794. [Google Scholar] [CrossRef]
- Duchêne, L.; Kim, D.H.; Song, Y.B.; Jun, S.; Moury, R.; Remhof, A.; Hagemann, H.; Jung, Y.S.; Battaglia, C. Crystallization of closo-borate electrolytes from solution enabling infiltration into slurry-casted porous electrodes for all-solid-state batteries. Energy Storage Mater. 2020, 26, 543–549. [Google Scholar] [CrossRef]
- Ezhov, A.V.; Vyal’ba, F.Y.; Kluykin, I.N.; Zhdanova, K.A.; Bragina, N.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Mironov, A.F.; Kuznetsov, N.T. Synthesis of New Bioinorganic Systems Based on Nitrilium Derivatives of closo-Decaborate Anion and meso-Arylporphyrins with Pendant Amino Groups. Macroheterocycles 2017, 10, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Mindich, A.L.; Bokach, N.A.; Kuznetsov, M.L.; Starova, G.L.; Zhdanov, A.P.; Zhizhin, K.Y.; Miltsov, S.A.; Kuznetsov, N.T.; Kukushkin, V.Y. Borylated Tetrazoles from Cycloaddition of Azide Anions to Nitrilium Derivatives of closo-Decaborate Clusters. Organometallics 2013, 32, 6576–6586. [Google Scholar] [CrossRef]
- Mindich, A.L.; Bokach, N.A.; Kuznetsov, M.L.; Haukka, M.; Zhdanov, A.P.; Zhizhin, K.Y.; Miltsov, S.A.; Kuznetsov, N.T.; Kukushkin, V.Y. Coupling of azomethine ylides with nitrilium derivatives of closo-decaborate clusters: A synthetic and theoretical study. ChemPlusChem 2012, 77, 1075–1086. [Google Scholar] [CrossRef]
- Mindich, A.L.; Bokach, N.A.; Dolgushin, F.M.; Haukka, M.; Lisitsyn, L.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Miltsov, S.A.; Kuznetsov, N.T.; Kukushkin, V.Y. 1,3-Dipolar cycloaddition of nitrones to a nitrile functionality in closo-decaborate clusters: A novel reactivity mode for the borylated C≡N group. Organometallics 2012, 31, 1716–1724. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Burianova, V.K.; Novikov, A.S.; Demakova, M.Y.; Pretorius, C.; Mokolokolo, P.P.; Roodt, A.; Bokach, N.A.; Suslonov, V.V.; Zhdanov, A.P.; et al. Nucleophilicity of Oximes Based upon Addition to a Nitrilium closo-Decaborate Cluster. Organometallics 2016, 35, 3612–3623. [Google Scholar] [CrossRef]
- Burianova, V.K.; Mikherdov, A.S.; Bolotin, D.S.; Novikov, A.S.; Mokolokolo, P.P.; Roodt, A.; Boyarskiy, V.P.; Suslonov, V.V.; Zhdanov, A.P.; Zhizhin, K.Y.; et al. Electrophilicity of aliphatic nitrilium closo-decaborate clusters: Hyperconjugation provides an unexpected inverse reactivity order. J. Organomet. Chem. 2018, 870, 97–103. [Google Scholar] [CrossRef]
- Burianova, V.K.; Bolotin, D.S.; Mikherdov, A.S.; Novikov, A.S.; Mokolokolo, P.P.; Roodt, A.; Boyarskiy, V.P.; Dar’In, D.; Krasavin, M.; Suslonov, V.V.; et al. Mechanism of generation of: Closo-decaborato amidrazones. Intramolecular non-covalent B-H⋯π (Ph) interaction determines stabilization of the configuration around the amidrazone CN bond. New J. Chem. 2018, 42, 8693–8703. [Google Scholar] [CrossRef]
- Nelyubin, A.V.; Klyukin, I.N.; Novikov, A.S.; Zhdanov, A.P.; Grigoriev, M.S.; Zhizhin, K.Y.; Kuznetsov, N.T. Nucleophilic addition of amino acid esters to nitrilium derivatives of closo-decaborate anion. Mendeleev Commun. 2021, 31, 201–203. [Google Scholar] [CrossRef]
- Zhdanov, A.P.; Voinova, V.V.; Klyukin, I.N.; Kubasov, A.S.; Zhizhin, K.Y.; Kuznetsov, N.T. New Synthesis Method of N-Monosubstituted Ammonium-closo-Decaborates. J. Clust. Sci. 2019, 30, 1327–1333. [Google Scholar] [CrossRef]
- Zhdanov, A.P.; Bykov, A.Y.; Kubasov, A.S.; Polyakova, I.N.; Razgonyaeva, G.A.; Zhizhin, K.Y.; Kuznetsov, N.T. Hydrolysis of nitrilium derivatives of the closo-decaborate anion [2-B10H9(N≡CR)]− (R = CH3, C2H5, C(CH3)3, or C6H5). Russ. J. Inorg. Chem. 2017, 62, 468–475. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Z.; Chen, H.; Wang, Z.; Zhou, X.; Zhang, H. Progress in three-dimensional aromatic-like closo-dodecaborate. Coord. Chem. Rev. 2021, 444, 214042. [Google Scholar] [CrossRef]
- Messina, M.S.; Axtell, J.C.; Wang, Y.; Chong, P.; Wixtrom, A.I.; Kirlikovali, K.O.; Upton, B.M.; Hunter, B.M.; Shafaat, O.S.; Khan, S.I.; et al. Visible-Light-Induced Olefin Activation Using 3D Aromatic Boron-Rich Cluster Photooxidants. J. Am. Chem. Soc. 2016, 138, 6952–6955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axtell, J.C.; Saleh, L.M.A.; Qian, E.A.; Wixtrom, A.I.; Spokoyny, A.M. Synthesis and Applications of Perfunctionalized Boron Clusters. Inorg. Chem. 2018, 57, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Diab, M.; Mateo, A.; Al Cheikh, J.; Haouas, M.; Ranjbari, A.; Bourdreux, F.; Naoufal, D.; Cadot, E.; Bo, C.; Floquet, S. Unprecedented coupling reaction between two anionic species of a closo -decahydrodecaborate cluster and an Anderson-type polyoxometalate. Dalton Trans. 2020, 49, 4685–4689. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.; Hamlin, D.K.; Perry, M.A. Reagents for Astatination of Biomolecules. 3. Comparison of closo - Decaborate(2-) and closo - Dodecaborate(2-) Moieties as Reactive Groups for Labeling with Astatine-211. Bioconjugate Chem. 2009, 20, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilbur, D.S.; Chyan, M.K.; Hamlin, D.K.; Perry, M.A. Preparation and in vivo evaluation of radioiodinated closo-decaborate(2-) derivatives to identify structural components that provide low retention in tissues. Nucl. Med. Biol. 2010, 37, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semioshkin, A.A.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of polyhedral boron hydrides and their synthetic applications. Dalton Trans. 2008, 11, 977–992. [Google Scholar] [CrossRef]
- Semioshkin, A.; Laskova, J.; Wojtczak, B.; Andrysiak, A.; Godovikov, I.; Bregadze, V.; Lesnikowski, Z.J. Synthesis of closo-dodecaborate based nucleoside conjugates. J. Organomet. Chem. 2009, 694, 1375–1379. [Google Scholar] [CrossRef]
- Olid, D.; Núñez, R.; Viñas, C.; Teixidor, F. Methods to produce B-C, B-P, B-N and B-S bonds in boron clusters. Chem. Soc. Rev. 2013, 42, 3318–3336. [Google Scholar] [CrossRef]
- Duttwyler, S. Recent advances in B-H functionalization of icosahedral carboranes and boranes by transition metal catalysis. Pure Appl. Chem. 2018, 90, 733–744. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bruskin, A.B.; Nesterov, V.V.; Antipin, M.Y.; Bregadze, V.I.; Sjöberg, S. Synthesis of Schiff bases derived from the ammoniaundecahydro-closo-dodecaborate(1-) anion, [B12H11NH=CHR]−, and their Reduction into monosubstituted amines [B12H11NH2CH2R]−: A new route to water soluble agents for BNCT. Inorg. Chem. 1999, 38, 5887–5893. [Google Scholar] [CrossRef]
- Hamdaoui, M.; Varkhedkar, R.; Sun, J.; Fan, L.; Duttwyler, S. Recent advances in the selective functionalization of anionic icosahedral boranes and carboranes. In Synthetic Inorganic Chemistry; Elsevier B.V.: Amsterdam, The Netherlands, 2021; pp. 343–389. [Google Scholar]
- Zhang, Y.; Sun, Y.; Wang, T.; Liu, J.; Spingler, B.; Duttwyler, S. Synthesis and Structural Characterization of Amidine, Amide, Urea and Isocyanate Derivatives of the Amino-closo-dodecaborate Anion [B12 H11 NH3]−. Molecules 2018, 23, 3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Zaria, M.E.; Ban, H.S.; Nakamura, H. Boron-containing protoporphyrin ix derivatives and their modification for boron neutron capture therapy: Synthesis, characterization, and comparative in vitro toxicity evaluation. Chem. Eur. J. 2010, 16, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Grüner, B.; Bonnetot, B.; Mongeot, H. Synthesis of N- and B-Substituted Derivatives of closo-Amino-undecahydro-dodecaborate(1-) Anion. Collect. Czechoslov. Chem. Commun. 1997, 62, 1185–1204. [Google Scholar] [CrossRef]
- Nelyubin, A.V.; Klyukin, I.N.; Zhdanov, A.P.; Grigor’ev, M.S.; Zhizhin, K.Y.; Kuznetsov, N.T. Synthesis of Nitrile Derivatives of the closo-Decaborate and closo-Dodecaborate Anions [BnHn−1NCR]− (n = 10, 12) by a Microwave Method. Russ. J. Inorg. Chem. 2021, 66, 139–145. [Google Scholar] [CrossRef]
- Zhdanov, A.P.; Polyakova, I.N.; Razgonyaeva, G.A.; Zhizhin, K.Y.; Kuznetsov, N.T. Reactions of nucleophilic addition of primary amines to the nitrilium derivative of the closo-decaborate anion [2-B10H9(N≡CCH3)]−. Russ. J. Inorg. Chem. 2011, 56, 847–855. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Dukenbayev, K.; Korolkov, I.; Tishkevich, D.; Kozlovskiy, A.; Trukhanov, S.; Gorin, Y.; Shumskaya, E.; Kaniukov, E.; Vinnik, D.; Zdorovets, M.; et al. Fe3O4 Nanoparticles for Complex Targeted Delivery and Boron Neutron Capture Therapy. Nanomaterials 2019, 9, 494. [Google Scholar] [CrossRef] [Green Version]
- Vorobyeva, M.A.; Dymova, M.A.; Novopashina, D.S.; Kuligina, E.V.; Timoshenko, V.V.; Kolesnikov, I.A.; Taskaev, S.Y.; Richter, V.A.; Venyaminova, A.G. Tumor Cell-Specific 2′-Fluoro RNA Aptamer Conjugated with Closo-Dodecaborate as A Potential Agent for Boron Neutron Capture Therapy. Int. J. Mol. Sci. 2021, 22, 7326. [Google Scholar] [CrossRef] [PubMed]
- Genady, A.R.; Ioppolo, J.A.; Azaam, M.M.; El-Zaria, M.E. New functionalized mercaptoundecahydrododecaborate derivatives for potential application in boron neutron capture therapy: Synthesis, characterization and dynamic visualization in cells. Eur. J. Med. Chem. 2015, 93, 574–583. [Google Scholar] [CrossRef]
- Koganei, H.; Tachikawa, S.; El-Zaria, M.E.; Nakamura, H. Synthesis of oligo-closo-dodecaborates by Hüisgen click reaction as encapsulated agents for the preparation of high-boron-content liposomes for neutron capture therapy. New J. Chem. 2015, 39, 6388–6394. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef]
- Losytskyy, M.Y.; Kovalska, V.B.; Varzatskii, O.A.; Kuperman, M.V.; Potocki, S.; Gumienna-Kontecka, E.; Zhdanov, A.P.; Yarmoluk, S.M.; Voloshin, Y.Z.; Zhizhin, K.Y.; et al. An interaction of the functionalized closo-borates with albumins: The protein fluorescence quenching and calorimetry study. J. Lumin. 2016, 169, 51–60. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- APEX2. Version 2.1-0; Bruker AXS Inc.: Madison, WI, USA, 2006.
- SADABS-2004/1; Bruker AXS Inc.: Fitchburg, WI, USA, 2004.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50, mM | ||||
|---|---|---|---|---|
| Cell Line | Na (3a) | Na (3c) | Na (3d) | Na (3g) |
| NKE | 3.27 ± 0.29 | 1.78 ± 0.25 | 1.09 ± 0.12 | 0.54 ± 0.08 |
| HaCat | 4.77 ± 0.87 | 4.43 ± 0.98 | 1.73 ± 0.20 | 1.20 ± 0.10 |
| U251 | 3.20 ± 0.47 | 4.10 ± 0.55 | 1.15 ± 0.28 | 0.54 ±0.03 |
| Hep2 | 6.55 ± 0.50 | 4.57 ± 0.74 | 2.33 ± 0.50 | 1.16 ± 0.11 |
| Dissociation Constant (Kd), 105 M−1 | ||||
|---|---|---|---|---|
| Peptide | Na (3a) | Na (3c) | Na (3d) | Na (3g) |
| BSA | 0.21 | 0.17 | 0.46 | 4.15 |
| HSA | 0.17 | 0.18 | 0.43 | 2.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelyubin, A.V.; Selivanov, N.A.; Bykov, A.Y.; Klyukin, I.N.; Novikov, A.S.; Zhdanov, A.P.; Karpechenko, N.Y.; Grigoriev, M.S.; Zhizhin, K.Y.; Kuznetsov, N.T. Primary Amine Nucleophilic Addition to Nitrilium Closo-Dodecaborate [B12H11NCCH3]−: A Simple and Effective Route to the New BNCT Drug Design. Int. J. Mol. Sci. 2021, 22, 13391. https://doi.org/10.3390/ijms222413391
Nelyubin AV, Selivanov NA, Bykov AY, Klyukin IN, Novikov AS, Zhdanov AP, Karpechenko NY, Grigoriev MS, Zhizhin KY, Kuznetsov NT. Primary Amine Nucleophilic Addition to Nitrilium Closo-Dodecaborate [B12H11NCCH3]−: A Simple and Effective Route to the New BNCT Drug Design. International Journal of Molecular Sciences. 2021; 22(24):13391. https://doi.org/10.3390/ijms222413391
Chicago/Turabian StyleNelyubin, Alexey V., Nikita A. Selivanov, Alexander Yu. Bykov, Ilya N. Klyukin, Alexander S. Novikov, Andrey P. Zhdanov, Natalia Yu. Karpechenko, Mikhail S. Grigoriev, Konstantin Yu. Zhizhin, and Nikolay T. Kuznetsov. 2021. "Primary Amine Nucleophilic Addition to Nitrilium Closo-Dodecaborate [B12H11NCCH3]−: A Simple and Effective Route to the New BNCT Drug Design" International Journal of Molecular Sciences 22, no. 24: 13391. https://doi.org/10.3390/ijms222413391