
Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons

Abstract
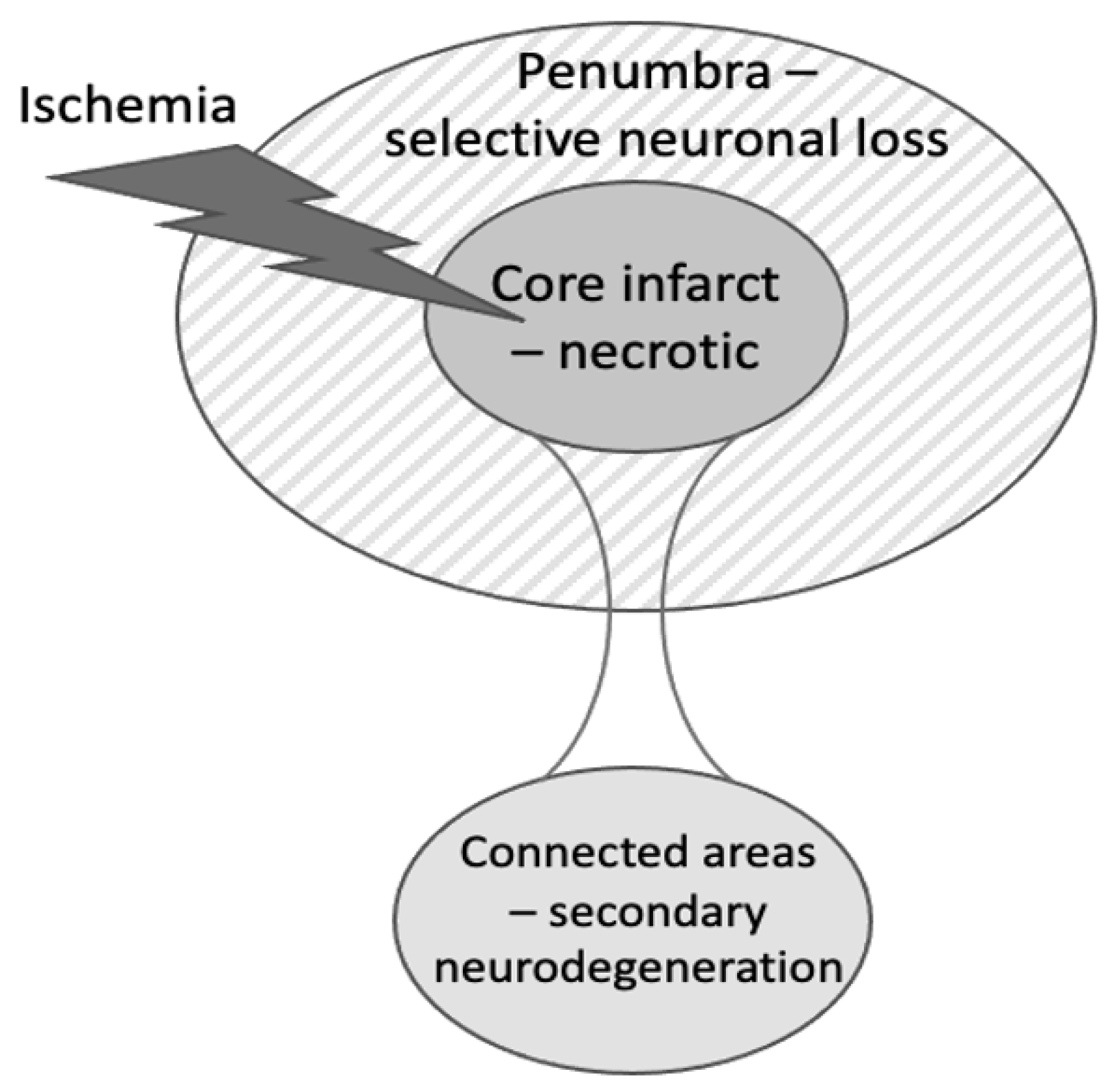
:1. Introduction
2. Types of Neuronal Death after Stroke
3. Microglia and Microglial Phagocytosis of Neurons
4. Phagocytic Signaling in Stroke
5. Evidence for Microglial Phagocytosis Causing Neuronal Loss in Peri-Infarct Areas after Stroke
6. Evidence for Microglial Phagocytosis in Secondary Neurodegeneration of the Thalamus after Stroke
7. Evidence for Microglial Phagocytosis in Delayed Neuronal Loss after Transient, Global Brain Ischemia
8. Evidence That Microglial Activation and Phagocytosis Are Beneficial after Stroke
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Sarmah, D.; Kalia, K.; Borah, A.; Wang, X.; Dave, K.R.; Yavagal, D.R.; Bhattacharya, P. Advances in Studies on Stroke-Induced Secondary Neurodegeneration (SND) and Its Treatment. Curr. Top. Med. Chem. 2020, 20, 1154–1168. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Tahira, Y.; Nagashima, H.; Kirino, T.; Gotoh, O.; Hojo, S.; Sano, K. Thalamic atrophy following cerebral infarction in the territory of the middle cerebral artery. Stroke 1991, 22, 615–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, J.C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982, 239, 57–69. [Google Scholar] [CrossRef]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef]
- Fujioka, M.; Taoka, T.; Matsuo, Y.; Mishima, K.; Ogoshi, K.; Kondo, Y.; Tsuda, M.; Fujiwara, M.; Asano, T.; Sakaki, T.; et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 2003, 54, 732–747. [Google Scholar] [CrossRef]
- Katchanov, J.; Waeber, C.; Gertz, K.; Gietz, A.; Winter, B.; Brück, W.; Dirnagl, U.; Veh, R.W.; Endres, M. Selective neuronal vulnerability following mild focal brain ischemia in the mouse. Brain Pathol. 2003, 13, 452–464. [Google Scholar] [CrossRef]
- Wegener, S.; Weber, R.; Ramos-Cabrer, P.; Uhlenkueken, U.; Sprenger, C.; Wiedermann, D.; Villringer, A.; Hoehn, M. Temporal profile of T2-weighted MRI distinguishes between pannecrosis and selective neuronal death after transient focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 2006, 26, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.H.; Liu, K.F.; Ye, Z.R.; Gutierrez, J.A. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke 1997, 28, 2303–2309. [Google Scholar] [CrossRef]
- Miyajima, N.; Ito, M.; Rokugawa, T.; Iimori, H.; Momosaki, S.; Omachi, S.; Shimosegawa, E.; Hatazawa, J.; Abe, K. Detection of neuroinflammation before selective neuronal loss appearance after mild focal ischemia using [18F]DPA-714 imaging. EJNMMI Res. 2018, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Emmrich, J.V.; Ejaz, S.; Williamson, D.J.; Hong, Y.T.; Sitnikov, S.; Fryer, T.D.; Aigbirhio, F.I.; Wulff, H.; Baron, J.C. Assessing the Effects of Cytoprotectants on Selective Neuronal Loss, Sensorimotor Deficit and Microglial Activation after Temporary Middle Cerebral Occlusion. Brain Sci. 2019, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Ejaz, S.; Williamson, D.J.; Ahmed, T.; Sitnikov, S.; Hong, Y.T.; Sawiak, S.J.; Fryer, T.D.; Aigbirhio, F.I.; Baron, J.C. Characterizing infarction and selective neuronal loss following temporary focal cerebral ischemia in the rat: A multi-modality imaging study. Neurobiol. Dis. 2013, 51, 120–132. [Google Scholar] [CrossRef]
- Cao, Z.; Harvey, S.S.; Bliss, T.M.; Cheng, M.Y.; Steinberg, G.K. Inflammatory Responses in the Secondary Thalamic Injury After Cortical Ischemic Stroke. Front. Neurol. 2020, 11, 236. [Google Scholar] [CrossRef]
- Sanchez-Bezanilla, S.; Hood, R.J.; Collins-Praino, L.E.; Turner, R.J.; Walker, F.R.; Nilsson, M.; Ong, L.K. More than motor impairment: A spatiotemporal analysis of cognitive impairment and associated neuropathological changes following cortical photothrombotic stroke. J. Cereb. Blood Flow Metab. 2021, 41, 2439–2455. [Google Scholar] [CrossRef]
- Forno, L.S. Reaction of the substantia nigra to massive basal ganglia infarction. Acta Neuropathol. 1983, 62, 96–102. [Google Scholar] [CrossRef]
- Ohara, S.; Kondo, K.; Kagoshima, M.; Yanagisawa, N. Secondary degeneration of substantia nigra following massive basal ganglia infarction. Rinsho Shinkeigaku 1989, 29, 1352–1356. [Google Scholar]
- Winter, B.; Juckel, G.; Viktorov, I.; Katchanov, J.; Gietz, A.; Sohr, R.; Balkaya, M.; Hörtnagl, H.; Endres, M. Anxious and hyperactive phenotype following brief ischemic episodes in mice. Biol. Psychiatry 2005, 57, 1166–1175. [Google Scholar] [CrossRef]
- Park, J.H.; Cho, J.H.; Ahn, J.H.; Choi, S.Y.; Lee, T.K.; Lee, J.C.; Shin, B.N.; Hong, S.; Jeon, Y.H.; Kim, Y.M.; et al. Neuronal loss and gliosis in the rat striatum subjected to 15 and 30 minutes of middle cerebral artery occlusion. Metab. Brain Dis. 2018, 33, 775–784. [Google Scholar] [CrossRef]
- Petito, C.K.; Feldmann, E.; Pulsinelli, W.A.; Plum, F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 1987, 37, 1281–1286. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem. Sci. 2012, 37, 325–332. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, J.; Zhang, Y.; Huang, Y.; Chen, D.; Shi, Z.; Smith, A.D.; Li, W.; Gao, Y. Central nervous system diseases related to pathological microglial phagocytosis. CNS Neurosci. Ther. 2021, 27, 528–539. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Savage, J.C.; Carrier, M.; Tremblay, M.È. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019, 2034, 13–26. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.R.; Choi, S.S.; Yeo, H.G.; Chang, K.T.; Lee, H.J. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed. Res. Int. 2014, 2014, 297241. [Google Scholar] [CrossRef]
- Kriz, J.; Lalancette-Hebert, M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009, 117, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, 1628, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.X.; Shen, H.; Huang, S.X.; Wang, L.P.; Li, Z.W.; Peng, P.; Mamtilahun, M.; Tang, Y.H.; Shen, F.X.; Tian, H.L.; et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P. Microglia as the Critical Regulators of Neuroprotection and Functional Recovery in Cerebral Ischemia. Cell Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Cockram, T.O.J.; Dundee, J.M.; Popescu, A.S.; Brown, G.C. The Phagocytic Code Regulating Phagocytosis of Mammalian Cells. Front. Immunol. 2021, 12, 629979. [Google Scholar] [CrossRef]
- Webster, C.M.; Hokari, M.; McManus, A.; Tang, X.N.; Ma, H.; Kacimi, R.; Yenari, M.A. Microglial P2Y12 deficiency/inhibition protects against brain ischemia. PLoS ONE 2013, 8, e70927. [Google Scholar] [CrossRef] [Green Version]
- Inose, Y.; Kato, Y.; Kitagawa, K.; Uchiyama, S.; Shibata, N. Activated microglia in ischemic stroke penumbra upregulate MCP-1 and CCR2 expression in response to lysophosphatidylcholine derived from adjacent neurons and astrocytes. Neuropathology 2015, 35, 209–223. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [Green Version]
- Soriano, S.G.; Amaravadi, L.S.; Wang, Y.F.; Zhou, H.; Yu, G.X.; Tonra, J.R.; Fairchild-Huntress, V.; Fang, Q.; Dunmore, J.H.; Huszar, D.; et al. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 2002, 125, 59–65. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Surugiu, R.; Catalin, B.; Dumbrava, D.; Gresita, A.; Olaru, D.G.; Hermann, D.M.; Popa-Wagner, A. Intracortical administration of the complement c3 receptor antagonist trifluoroacetate modulates microglia reaction after brain injury. Neural. Plast. 2019, 2019, 1071036. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-Y.; Pan, J.; Mamtilahun, M.; Zhu, Y.; Wang, L.; Venkatesh, A.; Shi, R.; Tu, X.; Jin, K.; Wang, Y.; et al. Microglia exacerbate white matter injury via complement c3/c3ar pathway after hypoperfusion. Theranostics 2020, 10, 74–90. [Google Scholar] [CrossRef]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [Green Version]
- Mari, C.; Karabiyikoglu, M.; Goris, M.L.; Tait, J.F.; Yenari, M.A.; Blankenberg, F.G. Detection of focal hypoxic-ischemic injury and neuronal stress in a rodent model of unilateral MCA occlusion/reperfusion using radiolabeled annexin V. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 733–739. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Li, X.; Wu, J.; Xue, T.; Wu, J.; Shen, H.; Li, X.; Shen, M.; Chen, G. TMEM16F Aggravates Neuronal Loss by Mediating Microglial Phagocytosis of Neurons in a Rat Experimental Cerebral Ischemia and Reperfusion Model. Front. Immunol. 2020, 11, 1144. [Google Scholar] [CrossRef]
- Neniskyte, U.; Brown, G.C. Lactadherin/MFG-E8 is essential for microglia-mediated neuronal loss and phagoptosis induced by amyloid β. J. Neurochem. 2013, 126, 312–317. [Google Scholar] [CrossRef]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Pedraza, C.E.; Nikolcheva, L.G.; Kaartinen, M.T.; Barralet, J.E.; McKee, M.D. Osteopontin functions as an opsonin and facilitates phagocytosis by macrophages of hydroxyapatite-coated microspheres: Implications for bone wound healing. Bone 2008, 43, 708–716. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, H.L.; Choi, J.S.; Choi, J.Y.; Cha, J.H.; Lee, M.Y. Osteopontin: Correlation with phagocytosis by brain macrophages in a rat model of stroke. Glia 2011, 59, 413–423. [Google Scholar] [CrossRef]
- Chung, A.G.; Frye, J.B.; Zbesko, J.C.; Constantopoulos, E.; Hayes, M.; Figueroa, A.G.; Becktel, D.A.; Antony Day, W.; Konhilas, J.P.; McKay, B.S.; et al. Liquefaction of the Brain following Stroke Shares a Similar Molecular and Morphological Profile with Atherosclerosis and Mediates Secondary Neurodegeneration in an Osteopontin-Dependent Mechanism. eNeuro 2018, 5, ENEURO.0076-18.2018. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Suzuma, K.; Otani, A.; Oh, H.; Koyama, S.; Ohashi, H.; Watanabe, D.; Ojima, T.; Suganami, E.; Honda, Y. Role of vitronectin receptor-type integrins and osteopontin in ischemia-induced retinal neovascularization. Jpn. J. Ophthalmol. 2002, 46, 270–278. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, H.L.; Park, J.M.; Cho, J.M.; Kim, C.Y.; Choi, K.J.; Kweon, H.S.; Cha, J.H.; Lee, M.Y. Overlapping distribution of osteopontin and calcium in the ischemic core of rat brain after transient focal ischemia. J. Neurotrauma 2012, 29, 1530–1538. [Google Scholar] [CrossRef]
- Ladwig, A.; Rogall, R.; Hucklenbroich, J.; Willuweit, A.; Schoeneck, M.; Langen, K.J.; Fink, G.R.; Rueger, M.A.; Schroeter, M. Osteopontin Attenuates Secondary Neurodegeneration in the Thalamus after Experimental Stroke. J. Neuroimmune Pharmacol. 2019, 14, 295–311. [Google Scholar] [CrossRef]
- Linnartz, B.; Kopatz, J.; Tenner, A.J.; Neumann, H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 2012, 32, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Kim, L.J.; Mealey, R.; Marsh, H.C., Jr.; Zhang, Y.; Tenner, A.J.; Connolly, E.S., Jr.; Pinsky, D.J. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science 1999, 285, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.; Xie, X.; Zhang, X.; Wang, Y.; Chu, K.; Brown, J.; Chen, L.; Hong, G. Inhibition of Complement Drives Increase in Early Growth Response Proteins and Neuroprotection Mediated by Salidroside After Cerebral Ischemia. Inflammation 2018, 41, 449–463. [Google Scholar] [CrossRef]
- Ten, V.S.; Sosunov, S.A.; Mazer, S.P.; Stark, R.I.; Caspersen, C.; Sughrue, M.E.; Botto, M.; Sander Connolly, E., Jr.; Pinsky, D.J. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke 2005, 36, 2244–2250. [Google Scholar] [CrossRef] [Green Version]
- Ten, V.S.; Yao, J.; Ratner, V.; Sosunov, S.; Fraser, D.A.; Botto, M.; Sivasankar, B.; Morgan, B.P.; Silverstein, S.; Stark, R.; et al. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J. Neurosci. 2010, 30, 2077–2087. [Google Scholar] [CrossRef] [Green Version]
- De Simoni, M.G.; Rossi, E.; Storini, C.; Pizzimenti, S.; Echart, C.; Bergamaschini, L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am. J. Pathol. 2004, 164, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Mocco, J.; Mack, W.J.; Ducruet, A.F.; Sosunov, S.A.; Sughrue, M.E.; Hassid, B.G.; Nair, N.M.; Laufer, I.; Komotar, R.J.; Claire, M.; et al. Complement Component C3 Mediates Inflammatory Injury Following Focal Cerebral Ischemia. Circ. Res. 2006, 99, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Alawieh, A.; Langley, E.F.; Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 2018, 10, eaao6459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawieh, A.M.; Langley, E.F.; Feng, W.; Spiotta, A.M.; Tomlinson, S. Complement-Dependent Synaptic Uptake and Cognitive Decline after Stroke and Reperfusion Therapy. J. Neurosci. 2020, 40, 4042–4058. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Q.; Lu, Y.; Dong, Y.; Dhandapani, K.M.; Brann, D.W.; Yu, R.K. Ganglioside GD3 is Up-regulated in Microglia and Regulates Phagocytosis Following Global Cerebral Ischemia. J. Neurochem. 2021, 158, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cao, L.L.; Wang, X.P.; Guo, W.; Guo, R.B.; Sun, Y.Q.; Xue, T.F.; Cai, Z.Y.; Ji, J.; Cheng, H.; et al. Neuronal extracellular vesicle derived miR-98 prevents salvageable neurons from microglial phagocytosis in acute ischemic stroke. Cell Death Dis. 2021, 12, 23. [Google Scholar] [CrossRef]
- Shi, X.; Luo, L.; Wang, J.; Shen, H.; Li, Y.; Mamtilahun, M.; Liu, C.; Shi, R.; Lee, J.H.; Tian, H.; et al. Stroke subtype-dependent synapse elimination by reactive gliosis in mice. Nat. Commun. 2021, 12, 6943. [Google Scholar] [CrossRef]
- Jin, Y.C.; Lee, H.; Wang, J.; Kim, S.W.; Kim, I.D.; Lee, H.K.; Lee, Y.; Han, P.L.; Lee, J.K. Intranasal Delivery of RGD Motif-Containing Osteopontin Icosamer Confers Neuroprotection in the Postischemic Brain via αvβ3 Integrin Binding. Mol. Neurobiol. 2016, 53, 5652–5663. [Google Scholar] [CrossRef]
- Ogawa, T.; Okudera, T.; Inugami, A.; Noguchi, K.; Kado, H.; Yoshida, Y.; Uemura, K. Degeneration of the ipsilateral substantia nigra after striatal infarction: Evaluation with MR imaging. Radiology 1997, 204, 847–851. [Google Scholar] [CrossRef]
- Kluge, M.G.; Abdolhoseini, M.; Zalewska, K.; Ong, L.K.; Johnson, S.J.; Nilsson, M.; Walker, F.R. Spatiotemporal analysis of impaired microglia process movement at sites of secondary neurodegeneration post-stroke. J. Cereb. Blood Flow Metab. 2019, 39, 2456–2470. [Google Scholar] [CrossRef]
- Holmberg, P.; Liljequist, S.; Wägner, A. Secondary brain injuries in thalamus and hippocampus after focal ischemia caused by mild, transient extradural compression of the somatosensori cortex in the rat. Curr. Neurovasc. Res. 2009, 6, 1–11. [Google Scholar] [CrossRef]
- Schaapsmeerders, P.; van Uden, I.W.; Tuladhar, A.M.; Maaijwee, N.A.; van Dijk, E.J.; Rutten-Jacobs, L.C.; Arntz, R.M.; Schoonderwaldt, H.C.; Dorresteijn, L.D.; de Leeuw, F.E.; et al. Ipsilateral hippocampal atrophy is associated with long-term memory dysfunction after ischemic stroke in young adults. Hum. Brain Mapp. 2015, 36, 2432–2442. [Google Scholar] [CrossRef]
- Hosp, J.A.; Greiner, K.L.; Martinez Arellano, L.; Roth, F.; Löffler, F.; Reis, J.; Fritsch, B. Progressive secondary exo-focal dopaminergic neurodegeneration occurs in not directly connected midbrain nuclei after pure motor-cortical stroke. Exp. Neurol. 2020, 327, 113211. [Google Scholar] [CrossRef]
- Pappata, S.; Levasseur, M.; Gunn, R.N.; Myers, R.; Crouzel, C.; Syrota, A.; Jones, T.; Kreutzberg, G.W.; Banati, R.B. Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]PK1195. Neurology 2000, 55, 1052–1054. [Google Scholar] [CrossRef]
- Schaechter, J.D.; Hightower, B.G.; Kim, M.; Loggia, M.L. A pilot [11C]PBR28 PET/MRI study of neuroinflammation and neurodegeneration in chronic stroke patients. Brain Behav. Immun. Health 2021, 17, 100336. [Google Scholar] [CrossRef]
- Jones, K.A.; Zouikr, I.; Patience, M.; Clarkson, A.N.; Isgaard, J.; Johnson, S.J.; Spratt, N.; Nilsson, M.; Walker, F.R. Chronic stress exacerbates neuronal loss associated with secondary neurodegeneration and suppresses microglial-like cells following focal motor cortex ischemia in the mouse. Brain Behav. Immun. 2015, 48, 57–67. [Google Scholar] [CrossRef]
- Cao, Z.; Harvey, S.S.; Chiang, T.; Foltz, A.G.; Lee, A.G.; Cheng, M.Y.; Steinberg, G.K. Unique Subtype of Microglia in Degenerative Thalamus After Cortical Stroke. Stroke 2021, 52, 687–698. [Google Scholar] [CrossRef]
- Zuo, X.; Hu, S.; Tang, Y.; Zhan, L.; Sun, W.; Zheng, J.; Han, Y.; Xu, E. Attenuation of secondary damage and Aβ deposits in the ipsilateral thalamus of dMCAO rats through reduction of cathepsin B by bis(propyl)-cognitin, a multifunctional dimer. Neuropharmacology 2020, 162, 107786. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.; Sunggip, C.; Tozaki-Saitoh, H.; Shimauchi, T.; Numaga-Tomita, T.; Hirano, K.; Ide, T.; Boeynaems, J.M.; Kurose, H.; Tsuda, M.; et al. Purinergic P2Y6 receptors heterodimerize with angiotensin AT1 receptors to promote angiotensin II-induced hypertension. Sci. Signal. 2016, 9, ra7. [Google Scholar] [CrossRef] [Green Version]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, C.; Hou, Z.; Fu, X.; Yuan, L.; Sun, S.; Zhang, H.; Yang, D.; Yao, X.; Yang, J. Pseudoginsenoside-F11 Accelerates Microglial Phagocytosis of Myelin Debris and Attenuates Cerebral Ischemic Injury Through Complement Receptor 3. Neuroscience 2020, 426, 33–49. [Google Scholar] [CrossRef]
- Cai, W.; Dai, X.; Chen, J.; Zhao, J.; Xu, M.; Zhang, L.; Yang, B.; Zhang, W.; Rocha, M.; Nakao, T.; et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 2019, 4, e131355. [Google Scholar] [CrossRef]
- Wu, R.; Li, X.; Xu, P.; Huang, L.; Cheng, J.; Huang, X.; Jiang, J.; Wu, L.J.; Tang, Y. TREM2 protects against cerebral ischemia/reperfusion injury. Mol. Brain. 2017, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Kurisu, K.; Zheng, Z.; Kim, J.Y.; Shi, J.; Kanoke, A.; Liu, J.; Hsieh, C.L.; Yenari, M.A. Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1906–1918. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Ji, J.; Sun, Y.; Huang, X.; Cai, Z.; Yang, J.; Guo, W.; Guo, R.; Cheng, H.; Sun, X. Sphingosine-1-phosphate, a novel TREM2 ligand, promotes microglial phagocytosis to protect against ischemic brain injury. Acta Pharm. Sin. B 2021. [Google Scholar] [CrossRef]
- Cheyuo, C.; Jacob, A.; Wu, R.; Zhou, M.; Qi, L.; Dong, W.; Ji, Y.; Chaung, W.W.; Wang, H.; Nicastro, J.; et al. Recombinant human MFG-E8 attenuates cerebral ischemic injury: Its role in anti-inflammation and anti-apoptosis. Neuropharmacology 2012, 62, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Deroide, N.; Li, X.; Lerouet, D.; Van Vré, E.; Baker, L.; Harrison, J.; Poittevin, M.; Masters, L.; Nih, L.; Margaill, I.; et al. MFGE8 inhibits inflammasome-induced IL-1β production and limits postischemic cerebral injury. J. Clin. Investig. 2013, 123, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Davaanyam, D.; Kim, I.D.; Lee, J.K. Intranasal Delivery of RGD-Containing Osteopontin Heptamer Peptide Confers Neuroprotection in the Ischemic Brain and Augments Microglia M2 Polarization. Int. J. Mol. Sci. 2021, 22, 9999. [Google Scholar] [CrossRef]
- Otxoa-de-Amezaga, A.; Miro-Mur, F.; Pedragosa, J.; Gallizioli, M.; Justicia, C.; Gaja-Capdevila, N.; Ruíz-Jaen, F.; Salas-Perdomo, A.; Bosch, A.; Calvo, M.; et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019, 137, 321–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Jiang, X.; Leak, R.K.; Shi, Y.; Hu, X.; Chen, J. Microglial Responses to Brain Injury and Disease: Functional Diversity and New Opportunities. Transl. Stroke Res. 2021, 12, 474–495. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Ref. | Stroke Model | Area of Neuron Loss | Time of Neuron Loss |
|---|---|---|---|
| [7] | 15 min MCAO in rats | Striatum | 0–16 weeks |
| [8] | 30 min MCAO in mice | Striatum | 0–6 weeks |
| [9] | 60 min MCAO in rats | Striatum | 0–6 weeks |
| [10] | 20 min MCAO in rats | Striatum and cortex | 1–7 days |
| [11] | 20 min MCAO in rats | Striatum | 1–7 days |
| [12] | 15 min MCAO in hypertensive rats | Cortex | 0–4 weeks |
| [13] | 45 min MCAO in hypertensive rats | Cortex | 0–4 weeks |
| [18] | 30 min MCAO in mice | Cortex | Unknown |
| [19] | 30 min MCAO in rats | Striatum | 0–5 days |
| [6] | Transient 4 vessel occlusion in rats | Cortex and hippocampus | 1–3 days |
| [15] | Photothrombosis in mouse cortex | Hippocampus | 7–84 days |
| [5] | 5 min carotid occlusion in gerbil | Hippocampal CA1 | 2–4 days |
| [3] | Ischemic stroke in human cortex | Thalamic atrophy by CT | 0–12 month |
| [16] | Basal ganglia stroke in humans | Substantia nigra | Unknown |
| [17] | MCAO in humans | Substantia nigra | Unknown |
| [20] | Cardiac arrest in humans | Hippocampus | 1–7 days |
| Ref. | Stroke Model | Intervention | Reduced Levels of |
|---|---|---|---|
| [43] | Transient, focal ischemia induced by endothelin 1 in mice | MFG-E8 knockout | Brain atrophy and motor deficits at 28 days |
| [43] | Transient, focal ischemia induced by endothelin 1 in rats | MerTK knockout | Brain atrophy and motor deficits at 28 days |
| [65] | Collagenase-induced intracerebral hemorrhage | MerTK knockout in microglia | Brain atrophy and motor deficits at 14 days |
| [65] | Transient MCAO in mice | MerTK knockout in microglia | Brain atrophy and motor deficits at 14 days |
| [66] | Transient MCAO in rats | RGD-peptides inhibiting αvβ3 | Infarct and motor deficits at 2 days |
| [45] | Transient MCAO in rats | TMEM16F knockdown | Neuron loss at 3 days Motor deficits 14 days |
| [64] | Transient MCAO in rats and mice | MicroRNA-98 overexpression | Neuron loss and neuro-deficits at unclear time |
| [55] | Transient MCAO in mice | C1q-blocking protein sCR1 | Neuro-deficits 1 day, infarct at 3 days |
| [60] | Transient MCAO in mice | C3 knockout | Infarct and neurological deficits at 1 day |
| [60] | Transient MCAO in mice | C3a-receptor antagonist | Infarct and neurological deficits at 1 day |
| [61] | Transient MCAO in mice | C3 inhibitor Crry | Neuron loss and neurodeficits at 15 days |
| [62] | Embolic MCAO in mice | C3 inhibitor Crry | Synapse loss and cognitive deficits at 30 days |
| [50] | dMCAO + hypoxia model of 2ry neurodegeneration in mice | Osteopontin knockout | Motor deficits 14 days, neuron loss at 49 days |
| [63] | Transient global brain ischemia in mice | GD3 synthase knockout | Neuron loss at 4 days |
| [36] | Transient global brain ischemia in mice | P2Y12 knockout or inhibition | Neuron loss at 3 days |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G.C. Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons. Int. J. Mol. Sci. 2021, 22, 13442. https://doi.org/10.3390/ijms222413442
Brown GC. Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons. International Journal of Molecular Sciences. 2021; 22(24):13442. https://doi.org/10.3390/ijms222413442
Chicago/Turabian StyleBrown, Guy C. 2021. "Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons" International Journal of Molecular Sciences 22, no. 24: 13442. https://doi.org/10.3390/ijms222413442
APA StyleBrown, G. C. (2021). Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons. International Journal of Molecular Sciences, 22(24), 13442. https://doi.org/10.3390/ijms222413442