
Cell Fate Decisions in the Neural Crest, from Pigment Cell to Neural Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neural Crest Development
2.1. Classical Views of Neural Crest Development
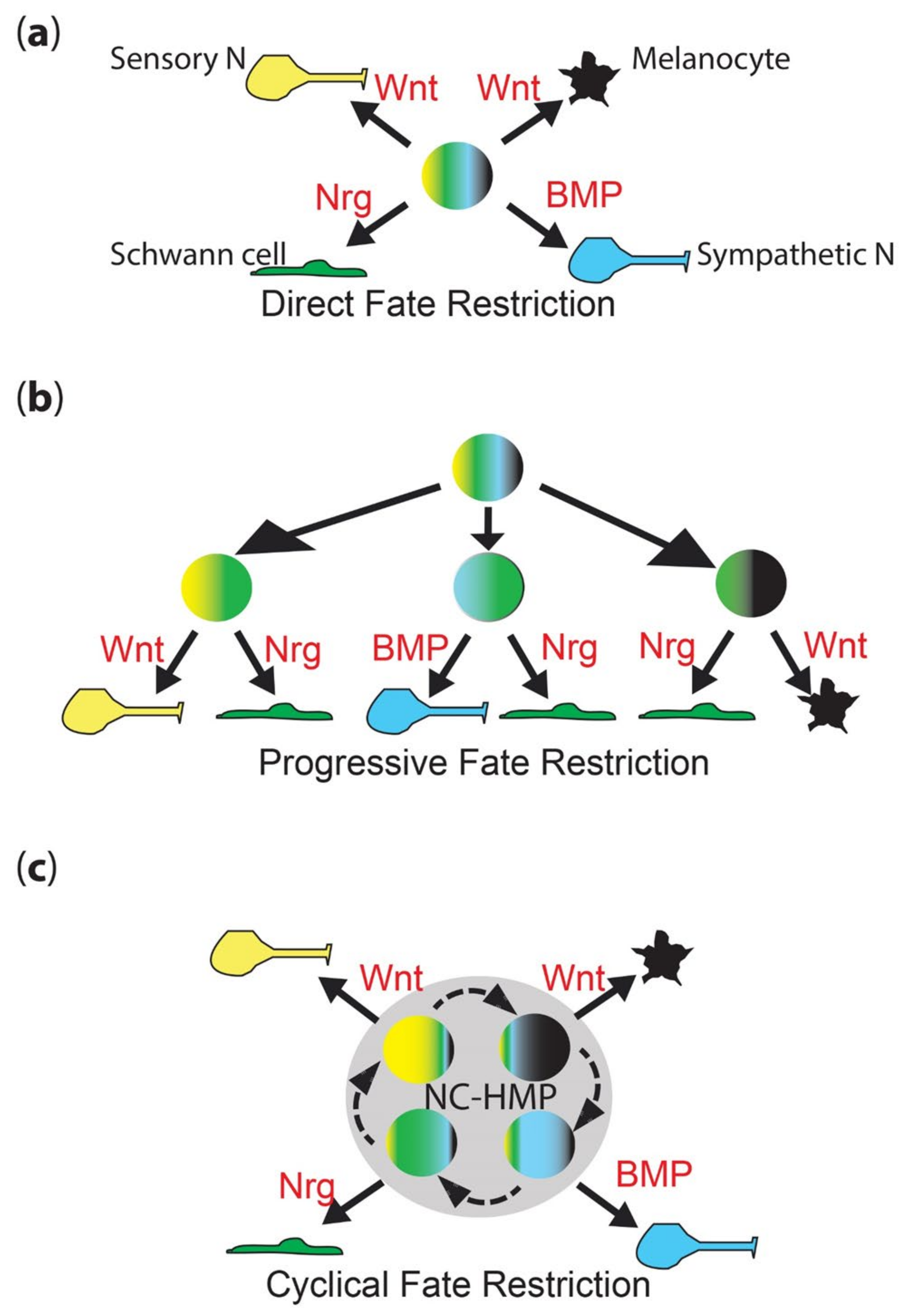
2.1.1. Direct Fate Restriction
2.1.2. Progressive Fate Restriction
2.2. Conflicting Observations within Recent Studies
2.2.1. Early Fate Specification and Ongoing Multipotency
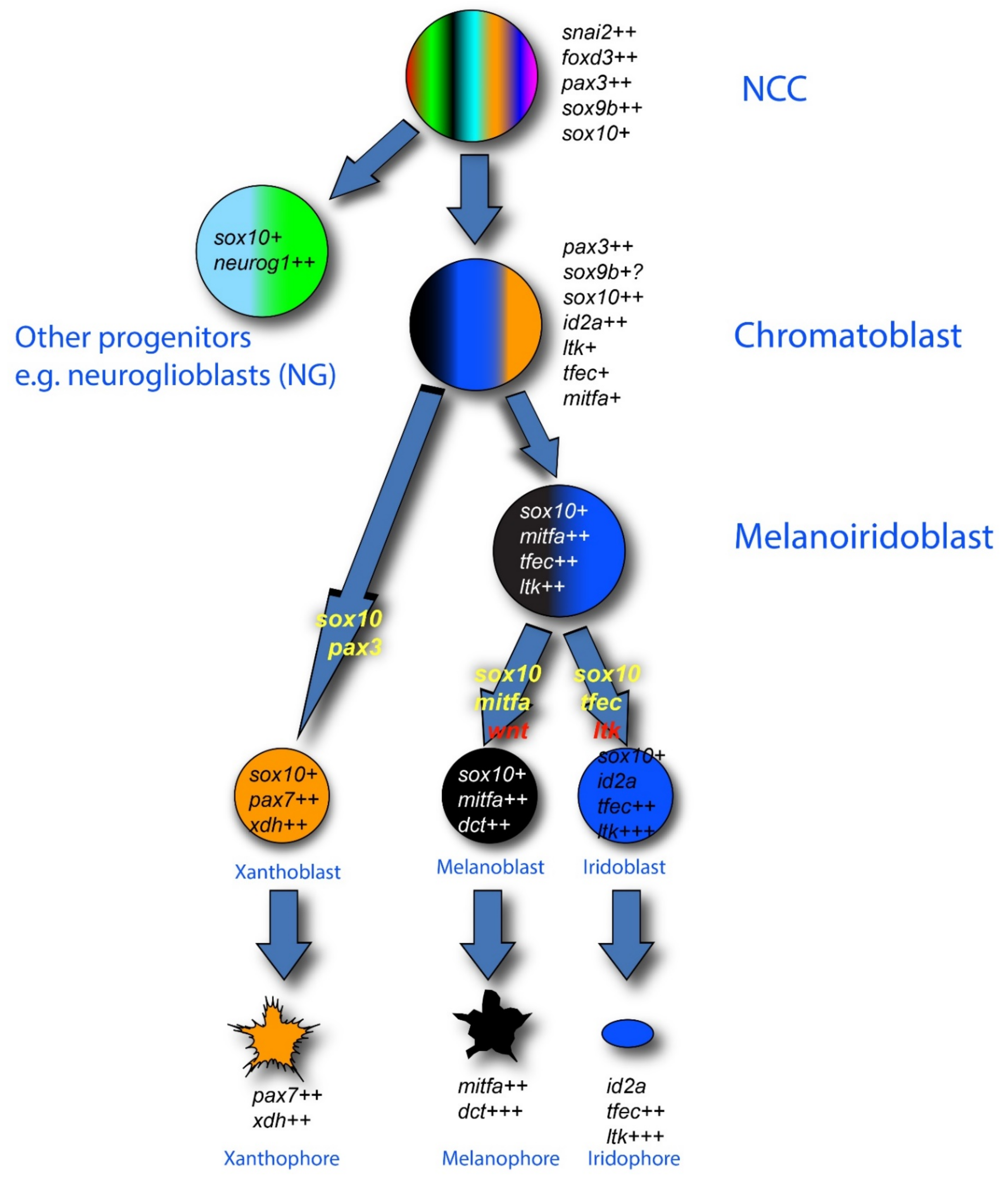
2.2.2. Zebrafish Pigment Cell Development
2.2.3. Broad Multipotency of Melanocyte Stem Cells
2.3. Resolving the Conflict?
2.3.1. Cyclical Fate Restriction
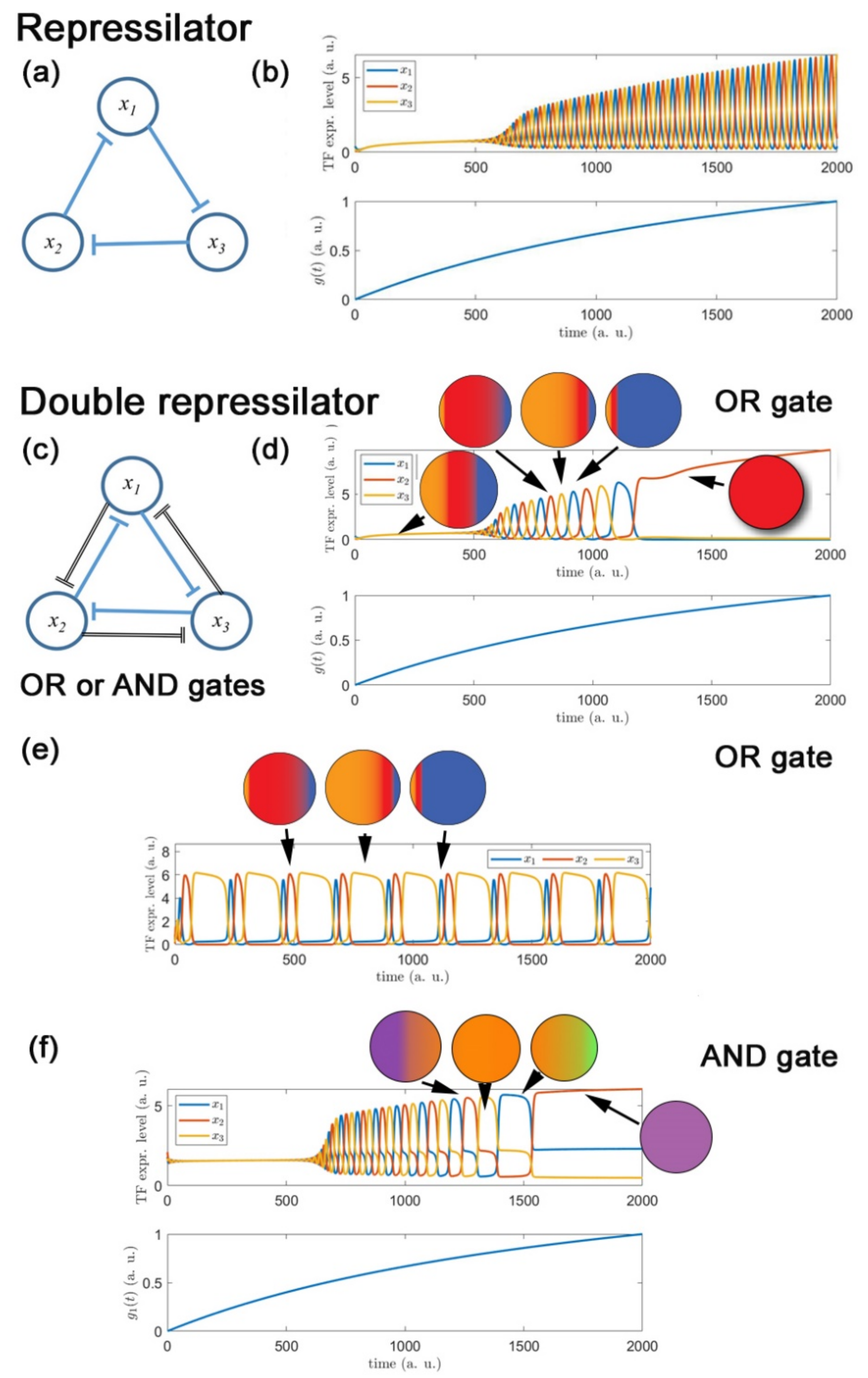
2.3.2. Dynamic Modelling of Fate Restriction Reproduces Key Features Consistent with Cyclical Fate Restriction
2.4. Implications and Further Work
2.4.1. Fate Specification to Multiple Fates Simultaneously
2.4.2. Cyclical Gene Expression
2.4.3. The Time Scale of Oscillations
2.4.4. When Does Commitment Occur?
2.4.5. Environmental Signals and NC-HMP Fate
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Le Douarin, N.M.; Kalcheim, C. The Neural Crest, 2nd ed.; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Dupin, E.; Baroffio, A.; Dulac, C.; Cameron-Curry, P.; Le Douarin, N. Schwann-cell differentiation in clonal cultures of the neural crest, as evidenced by the anti-Schwann cell myelin protein monoclonal antibody. Proc. Natl. Acad. Sci. USA 1990, 87, 1119–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieber-Blum, M.; Cohen, A.M. Clonal analysis of quail neural crest cells: They are pluripotent and differentiate in vitro in the absence of noncrest cells. Dev. Biol. 1980, 80, 96–106. [Google Scholar] [CrossRef]
- Sieber-Blum, M. Commitment of neural crest cells to the sensory neuron lineage. Science 1989, 243, 1608–1611. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Morita, T.; Sieber-Blum, M. In vitro clonal analysis of mouse neural crest development. Dev. Biol. 1993, 157, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Sieber-Blum, M. Pluripotent and developmentally restricted neural-crest-derived cells in posterior visceral arches. Dev. Biol. 1993, 156, 191–200. [Google Scholar] [CrossRef]
- Le Douarin, N.M. Cell line segregation during peripheral nervous system ontogeny. Science 1986, 231, 1515–1522. [Google Scholar] [CrossRef]
- Baroffio, A.; Dupin, E.; Le Douarin, N. Clone-forming ability and differentiation potential of migratory neural crest cells. Proc. Natl. Acad. Sci. USA 1988, 85, 5325–5329. [Google Scholar] [CrossRef] [Green Version]
- Baroffio, A.; Dupin, E.; Le Douarin, N.M. Common precursors for neural and mesectodermal derivatives in the cephalic neural crest. Development 1991, 112, 301–305. [Google Scholar] [CrossRef]
- Weston, J.A. Neural crest cell development. Prog. Clin. Biol. Res. 1982, 85 Pt B, 359–379. [Google Scholar]
- Weston, J.A. Regulation of neural crest cell migration and differentiation. In Cell Interactions and Development: Molecular Mechanisms; Yamada, K.M., Ed.; John Wiley and Sons, Inc: Hoboken, NJ, USA, 1983; pp. 153–184. [Google Scholar]
- Weston, J.A. Sequential Segregation and Fate of Developmentally Restricted Intermediate Cell Populations in the Neural Crest Lineage. Curr. Topics Dev. Biol. 1991, 25, 133–153. [Google Scholar]
- Bronner-Fraser, M.; Fraser, S.E. Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature 1988, 335, 161–164. [Google Scholar] [CrossRef]
- Bronner-Fraser, M.; Fraser, S. Developmental potential of avian trunk neural crest cells in situ. Neuron 1989, 3, 755–766. [Google Scholar] [CrossRef]
- Fraser, S.E.; Bronner-Fraser, M. Migrating neural crest cells in the trunk of the avian embryo are multipotent. Development 1991, 112, 913–920. [Google Scholar] [CrossRef]
- Nikaido, M.; Subkhankulova, T.; Kasianov, A.; Uroshlev, L.; Camargo Sosa, K.; Bavister, G.; Yang, X.; Rodrigues, F.S.L.M.; Carney, T.J.; Dawes, J.H.P.; et al. Zebrafish pigment cells develop directly from highly multipotent progenitors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kelsh, R.; Camargo-Sosa, K.; Farjami, S.; Makeev, V.; Dawes, J.; Rocco, A. Cyclical Fate Restriction, a new view of neural crest cell fate specification. Development 2021, 148, dev176057. [Google Scholar] [CrossRef]
- Stemple, D.L.; Anderson, D.J. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 1992, 71, 973–985. [Google Scholar] [CrossRef]
- Shah, N.M.; Marchionni, M.A.; Isaacs, I.; Stroobant, P.; Anderson, D.J. Glial growth-factor restricts mammalian neural crest stem-cells to a glial fate. Cell 1994, 77, 349–360. [Google Scholar] [CrossRef]
- Shah, N.M.; Groves, A.K.; Anderson, D.J. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell 1996, 85, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Lo, L.; Sommer, L.; Anderson, D.J. MASH1 maintains competence for BMP2-induced neuronal differentiation in post-migratory neural crest cells. Curr. Biol. 1997, 7, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.M.; Anderson, D.J. Integration of multiple instructive cues by neural crest stem cells reveals cell-intrinsic biases in relative growth factor responsiveness. Proc. Natl. Acad. Sci. USA 1997, 94, 11369–11374. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.E.; Rebelo, S.; Anderson, D.J. Early specification of sensory neuron fate revealed by expression and function of neurogenins in the chick embryo. Development 1999, 126, 1715–1728. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron 2003, 38, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Perez, S.E.; Qiao, Z.; Verdi, J.M.; Hicks, C.; Weinmaster, G.; Anderson, D.J. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell 2000, 101, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Reissmann, E.; Ernsberger, U.; Francis-West, P.; Rueger, D.; Brickell, P.; Rohrer, H. Involvement of bone morphogenetic protein-4 and bone morphogenetic protein-7 in the differentiation of the adrenergic phenotype in developing sympathetic neurons. Development 1996, 122, 2079–2088. [Google Scholar] [CrossRef]
- Hari, L.; Miescher, I.; Shakhova, O.; Suter, U.; Chin, L.; Taketo, M.; Richardson, W.D.; Kessaris, N.; Sommer, L. Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development 2012, 139, 2107–2117. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Vieceli, F.M.; Gonzalez, W.G.; Li, A.; Tang, W.; Lois, C.; Bronner, M.E. In Vivo Quantitative Imaging Provides Insights into Trunk Neural Crest Migration. Cell Rep. 2019, 26, 1489–1500.e1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, C.A.; Duong, T.D.; Tosney, K.W. Descriptive and experimental analysis of the dispersion of neural crest cells along the dorsolateral path and their entry into ectoderm in the chick embryo. Dev. Biol. 1992, 151, 251–272. [Google Scholar] [CrossRef] [Green Version]
- Reedy, M.V.; Faraco, C.D.; Erickson, C.A. The delayed entry of thoracic neural crest cells into the dorsolateral path is a consequence of the late emigration of melanogenic neural crest cells from the neural tube. Dev. Biol. 1998, 200, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krispin, S.; Nitzan, E.; Kassem, Y.; Kalcheim, C. Evidence for a dynamic spatiotemporal fate map and early fate restrictions of premigratory avian neural crest. Development 2010, 137, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzan, E.; Krispin, S.; Pfaltzgraff, E.R.; Klar, A.; Labosky, P.A.; Kalcheim, C. A dynamic code of dorsal neural tube genes regulates the segregation between neurogenic and melanogenic neural crest cells. Development 2013, 140, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzan, E.; Pfaltzgraff, E.R.; Labosky, P.A.; Kalcheim, C. Neural crest and Schwann cell progenitor-derived melanocytes are two spatially segregated populations similarly regulated by Foxd3. Proc. Natl. Acad. Sci. USA 2013, 110, 12709–12714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, K.; Takiguchi-Hayashi, K.; Sezaki, M.; Yamamoto, H.; Takeuchi, T. Avian neural crest cells express a melanogenic trait migration from the neural tube: Observations with the antibody, “MEBL-1”. Development 1992, 114, 367–378. [Google Scholar] [CrossRef]
- Serbedzija, G.N.; Bronner-Fraser, M.; Fraser, S.E. A vital dye analysis of the timing and pathways of avian trunk neural crest cell migration. Development 1989, 106, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F.; Barresi, M.J.F. Developmental Biology, 11th ed.; Sinauer Associates Inc.: Sunderland, MD, USA, 2016. [Google Scholar]
- Baggiolini, A.; Varum, S.; Mateos, J.M.; Bettosini, D.; John, N.; Bonalli, M.; Ziegler, U.; Dimou, L.; Clevers, H.; Furrer, R.; et al. Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem Cell 2015, 16, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldatov, R.; Kaucka, M.; Kastriti, M.E.; Petersen, J.; Chontorotzea, T.; Englmaier, L.; Akkuratova, N.; Yang, Y.; Haring, M.; Dyachuk, V.; et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364. [Google Scholar] [CrossRef] [Green Version]
- Ling, I.T.C.; Sauka-Spengler, T. Early chromatin shaping predetermines multipotent vagal neural crest into neural, neuronal and mesenchymal lineages. Nat. Cell Biol. 2019, 21, 1504–1517. [Google Scholar] [CrossRef]
- Kelsh, R.N.; Schmid, B.; Eisen, J.S. Genetic analysis of melanophore development in zebrafish embryos. Dev. Biol. 2000, 225, 277–293. [Google Scholar] [CrossRef]
- Lopes, S.S.; Yang, X.; Muller, J.; Carney, T.J.; McAdow, A.R.; Rauch, G.J.; Jacoby, A.S.; Hurst, L.D.; Delfino-Machin, M.; Haffter, P.; et al. Leukocyte tyrosine kinase functions in pigment cell development. PLoS Genet. 2008, 4, e1000026. [Google Scholar] [CrossRef] [Green Version]
- Petratou, K.; Subkhankulova, T.; Lister, J.A.; Rocco, A.; Schwetlick, H.; Kelsh, R.N. A systems biology approach uncovers the core gene regulatory network governing iridophore fate choice from the neural crest. PLoS Genet. 2018, 14, e1007402. [Google Scholar] [CrossRef]
- Petratou, K.; Spencer, S.A.; Kelsh, R.N.; Lister, J.A. The MITF paralog tfec is required in neural crest development for fate specification of the iridophore lineage from a multipotent pigment cell progenitor. PLoS ONE 2021, 16, e0244794. [Google Scholar] [CrossRef]
- Parichy, D.M.; Ransom, D.G.; Paw, B.; Zon, L.I.; Johnson, S.L. An orthologue of the kit-related gene fms is required for development of neural crest-derived xanthophores and a subpopulation of adult melanocytes in the zebrafish, Danio rerio. Development 2000, 127, 3031–3044. [Google Scholar] [CrossRef] [PubMed]
- Parichy, D.M.; Rawls, J.F.; Pratt, S.J.; Whitfield, T.T.; Johnson, S.L. Zebrafish sparse corresponds to an orthologue of c-kit and is required for the morphogenesis of a subpopulation of melanocytes, but is not essential for hematopoiesis or primordial germ cell development. Development 1999, 126, 3425–3436. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.; Raible, D.W.; Lister, J.A. Foxd3 controls melanophore specification in the zebrafish neural crest by regulation of Mitf. Dev. Biol. 2009, 332, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Lister, J.A.; Robertson, C.P.; Lepage, T.; Johnson, S.L.; Raible, D.W. nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development 1999, 126, 3757–3767. [Google Scholar] [CrossRef]
- Delfino-Machin, M.; Chipperfield, T.R.; Rodrigues, F.S.; Kelsh, R.N. The proliferating field of neural crest stem cells. Dev. Dyn. 2007, 236, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, E.K. Melanocyte stem cells: A melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment Cell Melanoma Res. 2011, 24, 401–410. [Google Scholar] [CrossRef]
- Watanabe, N.; Motohashi, T.; Nishioka, M.; Kawamura, N.; Hirobe, T.; Kunisada, T. Multipotency of melanoblasts isolated from murine skin depends on the Notch signal. Dev. Dyn. 2016, 245, 460–471. [Google Scholar] [CrossRef] [Green Version]
- McGraw, H.F.; Nechiporuk, A.; Raible, D.W. Zebrafish dorsal root ganglia neural precursor cells adopt a glial fate in the absence of neurogenin1. J. Neurosci. 2008, 28, 12558–12569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, T.J.; Dutton, K.A.; Greenhill, E.; Delfino-Machin, M.; Dufourcq, P.; Blader, P.; Kelsh, R.N. A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development 2006, 133, 4619–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, D.; Luther, W.; Wang, W.; Paw, B.H.; Stewart, R.A.; George, R.E. Distinct neuroblastoma-associated alterations of PHOX2B impair sympathetic neuronal differentiation in zebrafish models. PLoS Genet. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elworthy, S.; Pinto, J.P.; Pettifer, A.; Cancela, M.L.; Kelsh, R.N. Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mech. Dev. 2005, 122, 659–669. [Google Scholar] [CrossRef]
- Dutton, K.A.; Pauliny, A.; Lopes, S.S.; Elworthy, S.; Carney, T.J.; Rauch, J.; Geisler, R.; Haffter, P.; Kelsh, R.N. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 2001, 128, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Delfino-Machin, M.; Madelaine, R.; Busolin, G.; Nikaido, M.; Colanesi, S.; Camargo-Sosa, K.; Law, E.W.; Toppo, S.; Blader, P.; Tiso, N.; et al. Sox10 contributes to the balance of fate choice in dorsal root ganglion progenitors. PLoS ONE 2017, 12, e0172947. [Google Scholar] [CrossRef]
- Budi, E.H.; Patterson, L.B.; Parichy, D.M. Embryonic requirements for ErbB signaling in neural crest development and adult pigment pattern formation. Development 2008, 135, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honjo, Y.; Kniss, J.; Eisen, J.S. Neuregulin-mediated ErbB3 signaling is required for formation of zebrafish dorsal root ganglion neurons. Development 2008, 135, 2615–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffter, P.; Odenthal, J.; Mullins, M.C.; Lin, S.; Farrell, M.J.; Vogelsang, E.; Haas, F.; Brand, M.; van Eeden, F.J.; Furutani-Seiki, M.; et al. Mutations affecting pigmentation and shape of the adult zebrafish. Dev. Genes Evol. 1996, 206, 260–276. [Google Scholar] [CrossRef]
- Kelsh, R.N.; Brand, M.; Jiang, Y.J.; Heisenberg, C.P.; Lin, S.; Haffter, P.; Odenthal, J.; Mullins, M.C.; van Eeden, F.J.; Furutani-Seiki, M.; et al. Zebrafish pigmentation mutations and the processes of neural crest development. Development 1996, 123, 369–389. [Google Scholar] [CrossRef]
- Odenthal, J.; Rossnagel, K.; Haffter, P.; Kelsh, R.; Vogelsang, E.; Brand, M.; van Eeden, F.; Furutani-Seiki, M.; Granato, M.; Hammerschmidt, M.; et al. Mutations affecting xanthophore pigmentation in the zebrafish, Danio rerio. Development 1996, 123, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Lencer, E.; Prekeris, R.; Artinger, K.B. Single-cell RNA analysis identifies pre-migratory neural crest cells expressing markers of differentiated derivatives. Elife 2021, 10, e66078. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.L.; Africa, D.; Walker, C.; Weston, J.A. Genetic control of adult pigment stripe development in zebrafish. Dev. Biol. 1995, 167, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parichy, D.M.; Mellgren, E.M.; Rawls, J.F.; Lopes, S.S.; Kelsh, R.N.; Johnson, S.L. Mutational analysis of endothelin receptor b1 (rose) during neural crest and pigment pattern development in the zebrafish Danio rerio. Dev. Biol. 2000, 227, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Rawls, J.F.; Johnson, S.L. Zebrafish kit mutation reveals primary and secondary regulation of melanocyte development during fin stripe regeneration. Development 2000, 127, 3715–3724. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.L.; Nguyen, A.N.; Lister, J.A. Mitfa is required at multiple stages of melanocyte differentiation but not to establish the melanocyte stem cell. Dev. Biol. 2011, 350, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elworthy, S.; Lister, J.A.; Carney, T.J.; Raible, D.W.; Kelsh, R.N. Transcriptional regulation of mitfa accounts for the sox10 requirement in zebrafish melanophore development. Development 2003, 130, 2809–2818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schartl, M.; Larue, L.; Goda, M.; Bosenberg, M.W.; Hashimoto, H.; Kelsh, R.N. What is a vertebrate pigment cell? Pigment Cell Melanoma Res. 2016, 29, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raible, D.W.; Eisen, J.S. Restriction of neural crest cell fate in the trunk of the embryonic zebrafish. Development 1994, 120, 495–503. [Google Scholar] [CrossRef]
- Schilling, T.F.; Kimmel, C.B. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development 1994, 120, 483–494. [Google Scholar] [CrossRef]
- Minchin, J.E.; Hughes, S.M. Sequential actions of Pax3 and Pax7 drive xanthophore development in zebrafish neural crest. Dev. Biol. 2008, 317, 508–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, K.; Lister, J.A.; Kunkel, G.R.; Prendergast, A.; Parichy, D.M.; Raible, D.W. Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Dev. Biol. 2010, 344, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauss, J.; Frohnhofer, H.G.; Walderich, B.; Maischein, H.M.; Weiler, C.; Irion, U.; Nusslein-Volhard, C. Endothelin signalling in iridophore development and stripe pattern formation of zebrafish. Biol. Open 2014, 3, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadeev, A.; Krauss, J.; Singh, A.P.; Nusslein-Volhard, C. Zebrafish Leucocyte tyrosine kinase controls iridophore establishment, proliferation and survival. Pigment Cell Melanoma Res. 2016, 29, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Nord, H.; Dennhag, N.; Muck, J.; von Hofsten, J. Pax7 is required for establishment of the xanthophore lineage in zebrafish embryos. Mol. Biol. Cell 2016, 27, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Fadeev, A.; Mendoza-Garcia, P.; Irion, U.; Guan, J.; Pfeifer, K.; Wiessner, S.; Serluca, F.; Singh, A.P.; Nusslein-Volhard, C.; Palmer, R.H. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. USA 2018, 115, E630–E638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnara, J.T.; Matsumoto, J.; Ferris, W.; Frost, S.K.; Turner, W.A., Jr.; Tchen, T.T.; Taylor, J.D. Common origin of pigment cells. Science 1979, 203, 410–415. [Google Scholar] [CrossRef]
- Kelsh, R.N.; Inoue, C.; Momoi, A.; Kondoh, H.; Furutani-Seiki, M.; Ozato, K.; Wakamatsu, Y. The Tomita collection of medaka pigmentation mutants as a resource for understanding neural crest cell development. Mech. Dev. 2004, 121, 841–859. [Google Scholar] [CrossRef]
- Kimura, T.; Nagao, Y.; Hashimoto, H.; Yamamoto-Shiraishi, Y.; Yamamoto, S.; Yabe, T.; Takada, S.; Kinoshita, M.; Kuroiwa, A.; Naruse, K. Leucophores are similar to xanthophores in their specification and differentiation processes in medaka. Proc. Natl. Acad. Sci. USA 2014, 111, 7343–7348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, Y.; Suzuki, T.; Shimizu, A.; Kimura, T.; Seki, R.; Adachi, T.; Inoue, C.; Omae, Y.; Kamei, Y.; Hara, I.; et al. Sox5 functions as a fate switch in medaka pigment cell development. PLoS Genet. 2014, 10, e1004246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, L.M.; Mishra, A.K.; Aman, A.J.; Lewis, V.M.; Toomey, M.B.; Packer, J.S.; Qiu, X.; McFaline-Figueroa, J.L.; Corbo, J.C.; Trapnell, C.; et al. Thyroid hormone regulates distinct paths to maturation in pigment cell lineages. Elife 2019, 8, e45181. [Google Scholar] [CrossRef]
- Howard, A.G.T.; Baker, P.A.; Ibarra-Garcia-Padilla, R.; Moore, J.A.; Rivas, L.J.; Tallman, J.J.; Singleton, E.W.; Westheimer, J.L.; Corteguera, J.A.; Uribe, R.A. An atlas of neural crest lineages along the posterior developing zebrafish at single-cell resolution. Elife 2021, 10, e60005. [Google Scholar] [CrossRef]
- Rodrigues, F.S.; Doughton, G.; Yang, B.; Kelsh, R.N. A novel transgenic line using the Cre-lox system to allow permanent lineage-labeling of the zebrafish neural crest. Genesis 2012, 50, 750–757. [Google Scholar] [CrossRef]
- Green, S.A.; Uy, B.R.; Bronner, M.E. Ancient evolutionary origin of vertebrate enteric neurons from trunk-derived neural crest. Nature 2017, 544, 88–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Medina, I.; Jevans, B.; Boismoreau, F.; Chettouh, Z.; Enomoto, H.; Muller, T.; Birchmeier, C.; Burns, A.J.; Brunet, J.F. Dual origin of enteric neurons in vagal Schwann cell precursors and the sympathetic neural crest. Proc. Natl. Acad. Sci. USA 2017, 114, 11980–11985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adameyko, I.; Lallemend, F.; Aquino, J.B.; Pereira, J.A.; Topilko, P.; Muller, T.; Fritz, N.; Beljajeva, A.; Mochii, M.; Liste, I.; et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009, 139, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Adameyko, I.; Lallemend, F.; Furlan, A.; Zinin, N.; Aranda, S.; Kitambi, S.S.; Blanchart, A.; Favaro, R.; Nicolis, S.; Lubke, M.; et al. Sox2 and Mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 2012, 139, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Furlan, A.; Adameyko, I. Schwann cell precursor: A neural crest cell in disguise? Dev. Biol. 2018, 444, S25–S35. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Solovieva, T.; Bronner, M. Schwann cell precursors: Where they come from and where they go. Cells Dev. 2021, 166, 203686. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hou, L. Regulation of melanocyte stem cell behavior by the niche microenvironment. Pigment Cell Melanoma Res. 2018, 31, 556–569. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Chuong, C.M.; Lei, M. Regulation of melanocyte stem cells in the pigmentation of skin and its appendages: Biological patterning and therapeutic potentials. Exp. Dermatol. 2019, 28, 395–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnheiter, H.; Debbache, J. Development of Melanin-Bearing Pigment Cells in Birds and Mammals. In Pigments, Pigment Cells and Pigment Patterns; Hashimoto, H., Goda, M., Futahashi, R., Kelsh, R., Akiyama, T., Eds.; Springer Nature Singapore Pte Ltd.: Singapore, 2021; pp. 185–208. [Google Scholar]
- Sieber-Blum, M. Human epidermal neural crest stem cells as candidates for cell-based therapies, disease modeling, and drug discovery. Birth Defects Res. C Embryo Today 2014, 102, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.A.; Cheung, M. Neural crest stem cells and their potential therapeutic applications. Dev. Biol. 2016, 419, 199–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadasi Boroujeni, S.; Koontz, A.; Tseropoulos, G.; Kerosuo, L.; Mehrotra, P.; Bajpai, V.K.; Selvam, S.R.; Lei, P.; Bronner, M.E.; Andreadis, S.T. Neural crest stem cells from human epidermis of aged donors maintain their multipotency in vitro and in vivo. Sci. Rep. 2019, 9, 9750. [Google Scholar] [CrossRef] [PubMed]
- White, P.M.; Morrison, S.J.; Orimoto, K.; Kubu, C.J.; Verdi, J.M.; Anderson, D.J. Neural crest stem cells undergo cell-intrinsic developmental changes in sensitivity to instructive differentiation signals. Neuron 2001, 29, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Parichy, D.M.; Liang, Y. Evolution of Pigment Patterning in Teleosts. In Pigments, Pigment Cells and Pigment Patterns; Hashimoto, H., Goda, M., Futahashi, R., Kelsh, R., Akiyama, T., Eds.; Springer Nature Singapore Pte Ltd.: Singapore, 2021; pp. 309–342. [Google Scholar]
- Owen, J.; Yates, C.; Kelsh, R.N. Pigment Patterning in Teleosts. In Pigments, Pigment Cells and Pigment Patterns; Hashimoto, H., Goda, M., Futahashi, R., Kelsh, R., Akiyama, T., Eds.; Springer Nature Singapore Pte Ltd.: Singapore, 2021; pp. 247–292. [Google Scholar]
- Parichy, D.M. Evolution of pigment cells and patterns: Recent insights from teleost fishes. Curr. Opin. Genet. Dev. 2021, 69, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Parichy, D.M. Advancing biology through a deeper understanding of zebrafish ecology and evolution. Elife 2015, 4, e05635. [Google Scholar] [CrossRef] [PubMed]
- Engeszer, R.E.; Barbiano, L.A.; Ryan, M.J.; Parichy, D.M. Timing and plasticity of shoaling behaviour in the zebrafish, Danio rerio. Anim. Behav. 2007, 74, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Irion, U.; Singh, A.P.; Nusslein-Volhard, C. The Developmental Genetics of Vertebrate Color Pattern Formation: Lessons from Zebrafish. Curr. Top. Dev. Biol. 2016, 117, 141–169. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.T.; Sengelmann, R.D.; Johnson, S.L. Larval melanocyte regeneration following laser ablation in zebrafish. J. Investig. Dermatol. 2004, 123, 924–929. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.T.; Johnson, S.L. Small molecule-induced ablation and subsequent regeneration of larval zebrafish melanocytes. Development 2006, 133, 3563–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultman, K.A.; Budi, E.H.; Teasley, D.C.; Gottlieb, A.Y.; Parichy, D.M.; Johnson, S.L. Defects in ErbB-dependent establishment of adult melanocyte stem cells reveal independent origins for embryonic and regeneration melanocytes. PLoS Genet. 2009, 5, e1000544. [Google Scholar] [CrossRef] [Green Version]
- Hultman, K.A.; Johnson, S.L. Differential contribution of direct-developing and stem cell-derived melanocytes to the zebrafish larval pigment pattern. Dev. Biol. 2010, 337, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryon, R.C.; Higdon, C.W.; Johnson, S.L. Lineage relationship of direct-developing melanocytes and melanocyte stem cells in the zebrafish. PLoS ONE 2011, 6, e21010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parichy, D.M.; Spiewak, J.E. Origins of adult pigmentation: Diversity in pigment stem cell lineages and implications for pattern evolution. Pigment Cell Melanoma Res. 2015, 28, 31–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMenamin, S.K.; Bain, E.J.; McCann, A.E.; Patterson, L.B.; Eom, D.S.; Waller, Z.P.; Hamill, J.C.; Kuhlman, J.A.; Eisen, J.S.; Parichy, D.M. Thyroid hormone-dependent adult pigment cell lineage and pattern in zebrafish. Science 2014, 345, 1358–1361. [Google Scholar] [CrossRef] [Green Version]
- Mahalwar, P.; Walderich, B.; Singh, A.P.; Nusslein-Volhard, C. Local reorganization of xanthophores fine-tunes and colors the striped pattern of zebrafish. Science 2014, 345, 1362–1364. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Patterson, L.B.; Parichy, D.M. Post-embryonic nerve-associated precursors to adult pigment cells: Genetic requirements and dynamics of morphogenesis and differentiation. PLoS Genet. 2011, 7, e1002044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, C.M.; Mongera, A.; Walderich, B.; Nusslein-Volhard, C. On the embryonic origin of adult melanophores: The role of ErbB and Kit signalling in establishing melanophore stem cells in zebrafish. Development 2013, 140, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo-Sosa, K.; Colanesi, S.; Muller, J.; Schulte-Merker, S.; Stemple, D.; Patton, E.E.; Kelsh, R.N. Endothelin receptor Aa regulates proliferation and differentiation of Erb-dependent pigment progenitors in zebrafish. PLoS Genet. 2019, 15, e1007941. [Google Scholar] [CrossRef] [Green Version]
- Kelsh, R.N.; Barsh, G.S. A nervous origin for fish stripes. PLoS Genet. 2011, 7, e1002081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adameyko, I.; Lallemend, F. Glial versus melanocyte cell fate choice: Schwann cell precursors as a cellular origin of melanocytes. Cell Mol. Life Sci. 2010, 67, 3037–3055. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Dinwiddie, A.; Mahalwar, P.; Schach, U.; Linker, C.; Irion, U.; Nusslein-Volhard, C. Pigment Cell Progenitors in Zebrafish Remain Multipotent through Metamorphosis. Dev. Cell 2016, 38, 316–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lewis, V.M.; Saunders, L.M.; Larson, T.A.; Bain, E.J.; Sturiale, S.L.; Gur, D.; Chowdhury, S.; Flynn, J.D.; Allen, M.C.; Deheyn, D.D.; et al. Fate plasticity and reprogramming in genetically distinct populations of Danio leucophores. Proc. Natl. Acad. Sci. USA 2019, 116, 11806–11811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farjami, S.; Camargo Sosa, K.; Dawes, J.H.P.; Kelsh, R.N.; Rocco, A. Novel generic models for differentiating stem cells reveal oscillatory mechanisms. J. R. Soc. Interface 2021, 18, 20210442. [Google Scholar] [CrossRef]
- Elowitz, M.B.; Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403, 335–338. [Google Scholar] [CrossRef]
- Chubb, J.R.; Ford, H.Z.; Antolovic, V. Distributed and centralized control during differentiation. Dev. Cell 2021, 56, 2142–2144. [Google Scholar] [CrossRef]
- Seberg, H.E.; Van Otterloo, E.; Loftus, S.K.; Liu, H.; Bonde, G.; Sompallae, R.; Gildea, D.E.; Santana, J.F.; Manak, J.R.; Pavan, W.J.; et al. TFAP2 paralogs regulate melanocyte differentiation in parallel with MITF. PLoS Genet. 2017, 13, e1006636. [Google Scholar] [CrossRef] [PubMed]
- Sauka-Spengler, T.; Meulemans, D.; Jones, M.; Bronner-Fraser, M. Ancient evolutionary origin of the neural crest gene regulatory network. Dev. Cell 2007, 13, 405–420. [Google Scholar] [CrossRef] [Green Version]
- De Croze, N.; Maczkowiak, F.; Monsoro-Burq, A.H. Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc. Natl. Acad. Sci. USA 2011, 108, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Plouhinec, J.L.; Roche, D.D.; Pegoraro, C.; Figueiredo, A.L.; Maczkowiak, F.; Brunet, L.J.; Milet, C.; Vert, J.P.; Pollet, N.; Harland, R.M.; et al. Pax3 and Zic1 trigger the early neural crest gene regulatory network by the direct activation of multiple key neural crest specifiers. Dev. Biol. 2014, 386, 461–472. [Google Scholar] [CrossRef]
- Simoes-Costa, M.; Tan-Cabugao, J.; Antoshechkin, I.; Sauka-Spengler, T.; Bronner, M.E. Transcriptome analysis reveals novel players in the cranial neural crest gene regulatory network. Genome Res. 2014, 24, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ferras, O.; Bernas, G.; Farnos, O.; Toure, A.M.; Souchkova, O.; Pilon, N. A direct role for murine Cdx proteins in the trunk neural crest gene regulatory network. Development 2016, 143, 1363–1374. [Google Scholar] [CrossRef] [Green Version]
- Hockman, D.; Chong-Morrison, V.; Green, S.A.; Gavriouchkina, D.; Candido-Ferreira, I.; Ling, I.T.C.; Williams, R.M.; Amemiya, C.T.; Smith, J.J.; Bronner, M.E.; et al. A genome-wide assessment of the ancestral neural crest gene regulatory network. Nat. Commun. 2019, 10, 4689. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Candido-Ferreira, I.; Repapi, E.; Gavriouchkina, D.; Senanayake, U.; Ling, I.T.C.; Telenius, J.; Taylor, S.; Hughes, J.; Sauka-Spengler, T. Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo. Dev. Cell 2019, 51, 255–276.e257. [Google Scholar] [CrossRef] [Green Version]
- Seal, S.; Monsoro-Burq, A.H. Insights Into the Early Gene Regulatory Network Controlling Neural Crest and Placode Fate Choices at the Neural Border. Front. Physiol. 2020, 11, 608812. [Google Scholar] [CrossRef]
- Varum, S.; Baggiolini, A.; Zurkirchen, L.; Atak, Z.K.; Cantu, C.; Marzorati, E.; Bossart, R.; Wouters, J.; Hausel, J.; Tuncer, E.; et al. Yin Yang 1 Orchestrates a Metabolic Program Required for Both Neural Crest Development and Melanoma Formation. Cell Stem Cell 2019, 24, 637–653.e639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong-Morrison, V.; Sauka-Spengler, T. The Cranial Neural Crest in a Multiomics Era. Front. Physiol. 2021, 12, 634440. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.S.; Sauka-Spengler, T.; LaBonne, C. Induction of the neural crest state: Control of stem cell attributes by gene regulatory, post-transcriptional and epigenetic interactions. Dev. Biol. 2012, 366, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancur, P.; Bronner-Fraser, M.; Sauka-Spengler, T. Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. USA 2010, 107, 3570–3575. [Google Scholar] [CrossRef] [Green Version]
- Martik, M.L.; Bronner, M.E. Regulatory Logic Underlying Diversification of the Neural Crest. Trends Genet. 2017, 33, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Nitzan, E.; Avraham, O.; Kahane, N.; Ofek, S.; Kumar, D.; Kalcheim, C. Dynamics of BMP and Hes1/Hairy1 signaling in the dorsal neural tube underlies the transition from neural crest to definitive roof plate. BMC Biol. 2016, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Shtukmaster, S.; Schier, M.C.; Huber, K.; Krispin, S.; Kalcheim, C.; Unsicker, K. Sympathetic neurons and chromaffin cells share a common progenitor in the neural crest in vivo. Neural Dev. 2013, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Greenhill, E.R.; Rocco, A.; Vibert, L.; Nikaido, M.; Kelsh, R.N. An iterative genetic and dynamical modelling approach identifies novel features of the gene regulatory network underlying melanocyte development. PLoS Genet. 2011, 7, e1002265. [Google Scholar] [CrossRef] [Green Version]
- Vibert, L.; Aquino, G.; Gehring, I.; Subkankulova, T.; Schilling, T.F.; Rocco, A.; Kelsh, R.N. An ongoing role for Wnt signaling in differentiating melanocytes in vivo. Pigment Cell Melanoma Res. 2016, 30, 219–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, Y.; Takada, H.; Miyadai, M.; Adachi, T.; Seki, R.; Kamei, Y.; Hara, I.; Taniguchi, Y.; Naruse, K.; Hibi, M.; et al. Distinct interactions of Sox5 and Sox10 in fate specification of pigment cells in medaka and zebrafish. PLoS Genet. 2018, 14, e1007260. [Google Scholar] [CrossRef]
- Van Otterloo, E.; Li, W.; Garnett, A.; Cattell, M.; Medeiros, D.M.; Cornell, R.A. Novel Tfap2-mediated control of soxE expression facilitated the evolutionary emergence of the neural crest. Development 2012, 139, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cornell, R.A. Redundant activities of Tfap2a and Tfap2c are required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev. Biol. 2007, 304, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Rothstein, M.; Azambuja, A.P.; Simoes-Costa, M. Control of neural crest multipotency by Wnt signaling and the Lin28/let-7 axis. Elife 2018, 7, e40556. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, M.; Simoes-Costa, M. Heterodimerization of TFAP2 pioneer factors drives epigenomic remodeling during neural crest specification. Genome Res. 2020, 30, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, A.P.; Simoes-Costa, M. The connectome of neural crest enhancers reveals regulatory features of signaling systems. Dev. Cell 2021, 56, 1268–1282.e1266. [Google Scholar] [CrossRef]
- Azambuja, A.P.; Simoes-Costa, M. A regulatory sub-circuit downstream of Wnt signaling controls developmental transitions in neural crest formation. PLoS Genet. 2021, 17, e1009296. [Google Scholar] [CrossRef] [PubMed]
- Ochi, S.; Imaizumi, Y.; Shimojo, H.; Miyachi, H.; Kageyama, R. Oscillatory expression of Hes1 regulates cell proliferation and neuronal differentiation in the embryonic brain. Development 2020, 147, dev182204. [Google Scholar] [CrossRef] [PubMed]
- Dorsky, R.I.; Moon, R.T.; Raible, D.W. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998, 396, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Dorsky, R.I.; Raible, D.W.; Moon, R.T. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 2000, 14, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.L.; Bonner, J.; Modrell, M.; Ragland, J.W.; Moon, R.T.; Dorsky, R.I.; Raible, D.W. Reiterated Wnt signaling during zebrafish neural crest development. Development 2004, 131, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenev, D.; Sunadome, K.; Shirokov, M.; Chagin, A.S.; Singh, A.; Irion, U.; Adameyko, I.; Fried, K.; Dyachuk, V. Schwann cell precursors generate sympathoadrenal system during zebrafish development. J. Neurosci. Res. 2021, 99, 2540–2557. [Google Scholar] [CrossRef] [PubMed]
- Kameneva, P.; Kastriti, M.E.; Adameyko, I. Neuronal lineages derived from the nerve-associated Schwann cell precursors. Cell Mol. Life Sci. 2021, 78, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Kaucka, M.; Szarowska, B.; Kavkova, M.; Kastriti, M.E.; Kameneva, P.; Schmidt, I.; Peskova, L.; Joven Araus, A.; Simon, A.; Kaiser, J.; et al. Nerve-associated Schwann cell precursors contribute extracutaneous melanocytes to the heart, inner ear, supraorbital locations and brain meninges. Cell Mol. Life Sci. 2021, 78, 6033–6049. [Google Scholar] [CrossRef]
- Moro, E.; Vettori, A.; Porazzi, P.; Schiavone, M.; Rampazzo, E.; Casari, A.; Ek, O.; Facchinello, N.; Astone, M.; Zancan, I.; et al. Generation and application of signaling pathway reporter lines in zebrafish. Mol. Genet. Genom. 2013, 288, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Casari, A.; Schiavone, M.; Facchinello, N.; Vettori, A.; Meyer, D.; Tiso, N.; Moro, E.; Argenton, F. A Smad3 transgenic reporter reveals TGF-beta control of zebrafish spinal cord development. Dev. Biol. 2014, 396, 81–93. [Google Scholar] [CrossRef]
- Astone, M.; Lai, J.K.H.; Dupont, S.; Stainier, D.Y.R.; Argenton, F.; Vettori, A. Zebrafish mutants and TEAD reporters reveal essential functions for Yap and Taz in posterior cardinal vein development. Sci. Rep. 2018, 8, 10189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, M.; Rampazzo, E.; Casari, A.; Battilana, G.; Persano, L.; Moro, E.; Liu, S.; Leach, S.D.; Tiso, N.; Argenton, F. Zebrafish reporter lines reveal in vivo signaling pathway activities involved in pancreatic cancer. Dis. Model. Mech. 2014, 7, 883–894. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawes, J.H.P.; Kelsh, R.N. Cell Fate Decisions in the Neural Crest, from Pigment Cell to Neural Development. Int. J. Mol. Sci. 2021, 22, 13531. https://doi.org/10.3390/ijms222413531
Dawes JHP, Kelsh RN. Cell Fate Decisions in the Neural Crest, from Pigment Cell to Neural Development. International Journal of Molecular Sciences. 2021; 22(24):13531. https://doi.org/10.3390/ijms222413531
Chicago/Turabian StyleDawes, Jonathan H. P., and Robert N. Kelsh. 2021. "Cell Fate Decisions in the Neural Crest, from Pigment Cell to Neural Development" International Journal of Molecular Sciences 22, no. 24: 13531. https://doi.org/10.3390/ijms222413531
APA StyleDawes, J. H. P., & Kelsh, R. N. (2021). Cell Fate Decisions in the Neural Crest, from Pigment Cell to Neural Development. International Journal of Molecular Sciences, 22(24), 13531. https://doi.org/10.3390/ijms222413531