
On the Road to Accurate Protein Biomarkers in Prostate Cancer Diagnosis and Prognosis: Current Status and Future Advances


Abstract
:1. Introduction
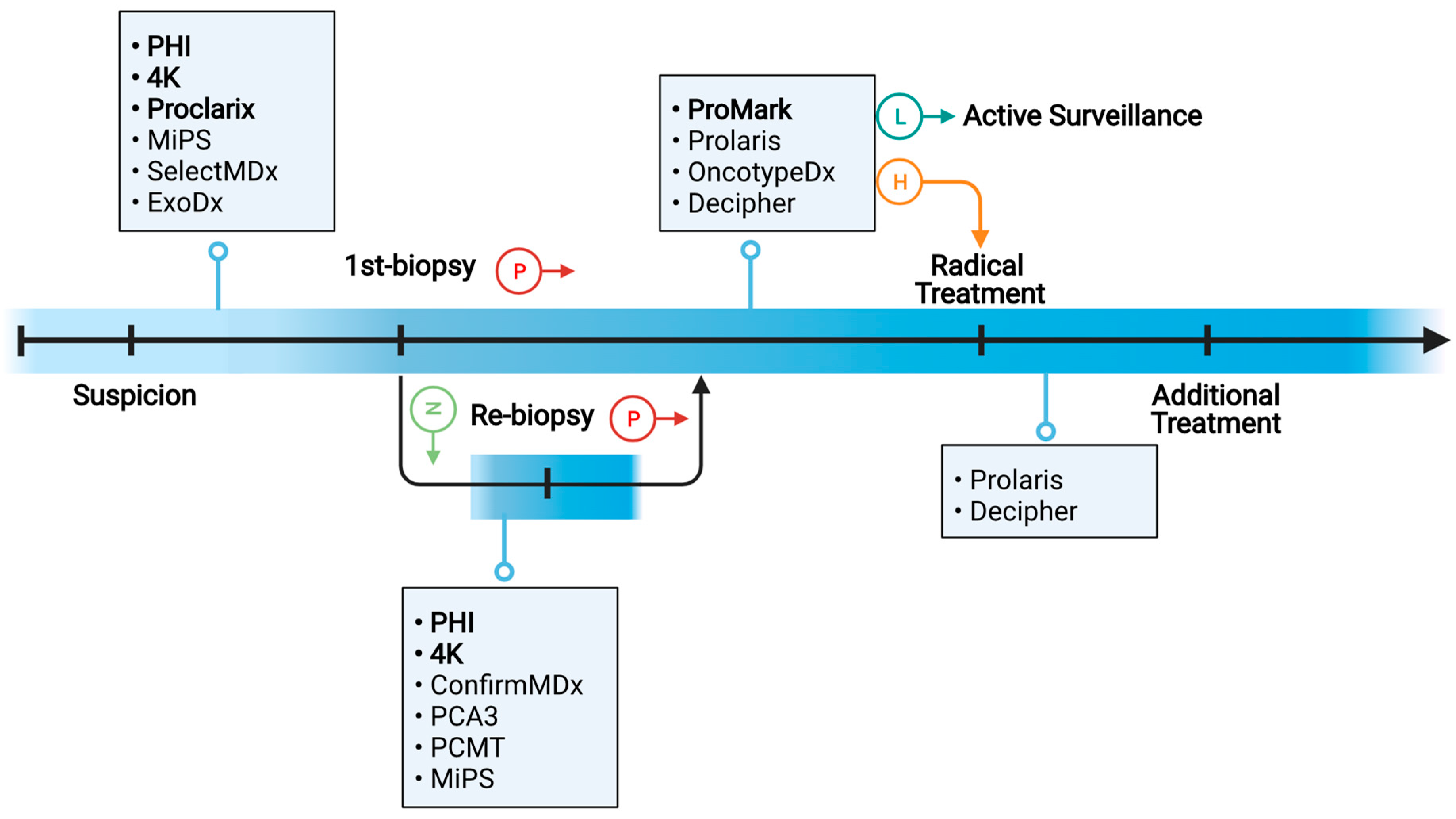
2. Clinical Needs of Molecular Biomarkers in PC Diagnosis and Prognosis
3. FDA-Approved and Commercially Available Protein Biomarkers for PC Diagnosis and Prognosis
4. Clinical Biospecimens for the Discovery and Validation of PC Protein Biomarkers
5. Proteomic Approaches for Global Discovery of Novel Protein Biomarkers from Clinical PC Specimens
5.1. MS-Based Protein Biomarker Discovery
5.2. Immunoassay-Based Protein Biomarker Discovery
5.2.1. Antibody Array
5.2.2. Antigen Array
5.2.3. Proximity Extension Assay (PEA)
5.3. Aptamer-Based Protein Biomarker Discovery
6. Computational Approaches for Prioritizing Protein Biomarker Candidates
7. Proteomic Approaches for Multiplexed Targeted Validation of Candidate PC Biomarker Proteins
7.1. MS-Based Multiplexed Targeted Proteomics
7.2. Antibody-Based Multiplexed Targeted Proteomics
7.2.1. RPPA
7.2.2. Microsphere Bead Capture (Luminex)
8. Challenges and Potential Solutions in Identifying and Validating Clinically Valuable PC Protein Biomarkers
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Department of Economic and Social Affairs, U.N. World Population Prospects 2019: Highlights. Dept. Econ. Soc. Aff. World Popul. Prospect. 2019, 46. [Google Scholar]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Prim. 2021, 7, 9. [Google Scholar] [CrossRef]
- Gleason, D.F. Classification of Prostatic Carcinomas. Cancer Chemother. Rep. 1966, 50, 125–128. [Google Scholar]
- Epstein, J.I. Prostate Cancer Grading: A Decade After the 2005 Modified System. Mod. Pathol. 2018, 31, 47–63. [Google Scholar] [CrossRef]
- Welch, H.G.; Fisher, E.S.; Gottlieb, D.J.; Barry, M.J. Detection of Prostate Cancer via Biopsy in the Medicare-SEER Population During the PSA Era. J. Natl. Cancer Inst. 2007, 99, 1395–1400. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Ioffe, V.; Huebner, T.; Hristova, I. Prostate Biopsy Characteristics: A Comparison Between the Pre- and Post-2012 United States Preventive Services Task Force (USPSTF) Prostate Cancer Screening Guidelines. Rev. Urol. 2018, 20, 77–83. [Google Scholar] [CrossRef]
- Schröder, F.H.; Hugosson, J.; Roobol, M.J.; Tammela, T.L.J.; Zappa, M.; Nelen, V.; Kwiatkowski, M.; Lujan, M.; Määttänen, L.; Lilja, H.; et al. Screening and Prostate Cancer Mortality: Results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of Follow-up. Lancet 2014, 384, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Loeb, S.; Vellekoop, A.; Ahmed, H.U.; Catto, J.; Emberton, M.; Nam, R.; Rosario, D.J.; Scattoni, V.; Lotan, Y. Systematic Review of Complications of Prostate Biopsy. Eur. Urol. 2013, 64, 876–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, W.; de Jong, H.; Melchers, W.; Mulders, P.; Schalken, J. Commercialized Blood-, Urinary- and Tissue-Based Biomarker Tests for Prostate Cancer Diagnosis and Prognosis. Cancers 2020, 12, 3790. [Google Scholar] [CrossRef]
- Meehan, J.; Gray, M.; Martínez-Pérez, C.; Kay, C.; McLaren, D.; Turnbull, A.K. Tissue- and Liquid-based Biomarkers in Prostate Cancer Precision Medicine. J. Pers. Med. 2021, 11, 664. [Google Scholar] [CrossRef]
- Capitanio, U.; Pfister, D.; Emberton, M. Repeat Prostate Biopsy: Rationale, Indications, and Strategies. Eur. Urol. Focus 2015, 1, 127–136. [Google Scholar] [CrossRef]
- Blute, M.L.; Abel, E.J.; Downs, T.M.; Kelcz, F.; Jarrard, D.F. Addressing the Need for Repeat Prostate Biopsy: New Technology and Approaches. Nat. Rev. Urol. 2015, 12, 435–444. [Google Scholar] [CrossRef]
- Liu, J.L.; Patel, H.D.; Haney, N.M.; Epstein, J.I.; Partin, A.W. Advances in the Selection of Patients with Prostate Cancer for Active Surveillance. Nat. Rev. Urol. 2021, 18, 197–208. [Google Scholar] [CrossRef]
- Dinh, K.T.; Mahal, B.A.; Ziehr, D.R.; Muralidhar, V.; Chen, Y.W.; Viswanathan, V.B.; Nezolosky, M.D.; Beard, C.J.; Choueiri, T.K.; Martin, N.E.; et al. Incidence and Predictors of Upgrading and Up Staging among 10,000 Contemporary Patients with Low Risk Prostate Cancer. J. Urol. 2015, 194, 343–349. [Google Scholar] [CrossRef]
- Wenzel, M.; Würnschimmel, C.; Chierigo, F.; Flammia, R.S.; Tian, Z.; Shariat, S.F.; Gallucci, M.; Terrone, C.; Saad, F.; Tilki, D.; et al. Nomogram Predicting Downgrading in National Comprehensive Cancer Network High-risk Prostate Cancer Patients Treated with Radical Prostatectomy. Eur. Urol. Focus 2021, 49, 1–8. [Google Scholar] [CrossRef]
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Cheng, H.H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T.; Eastham, J.A.; et al. Prostate Cancer, Version 1.2022, NCCN Clinical Practice Guidelines in Oncology. Natl. Compr. Cancer Netw. 2021. [Google Scholar]
- Ross, A.E.; Yousefi, K.; Davicioni, E.; Ghadessi, M.; Johnson, M.H.; Sundi, D.; Tosoian, J.J.; Han, M.; Humphreys, E.B.; Partin, A.W.; et al. Utility of Risk Models in Decision Making after Radical Prostatectomy: Lessons from a Natural History Cohort of Intermediate- and High-Risk Men. Eur. Urol. 2016, 69, 496–504. [Google Scholar] [CrossRef]
- Olleik, G.; Kassouf, W.; Aprikian, A.; Hu, J.; Vanhuyse, M.; Cury, F.; Peacock, S.; Bonnevier, E.; Palenius, E.; Dragomir, A. Evaluation of New Tests and Interventions for Prostate Cancer Management: A Systematic Review. JNCCN J. Natl. Compr. Cancer Netw. 2018, 16, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Couñago, F.; López-Campos, F.; Díaz-Gavela, A.A.; Almagro, E.; Fenández-Pascual, E.; Henríquez, I.; Lozano, R.; Espinós, E.L.; Gómez-Iturriaga, A.; de Velasco, G.; et al. Clinical Applications of Molecular Biomarkers in Prostate Cancer. Cancers 2020, 12, 1550. [Google Scholar] [CrossRef]
- Balázs, K.; Antal, L.; Sáfrány, G.; Lumniczky, K. Blood-derived Biomarkers of Diagnosis, Prognosis and Therapy Response in Prostate Cancer Patients. J. Pers. Med. 2021, 11, 296. [Google Scholar] [CrossRef]
- Eyrich, N.W.; Morgan, T.M.; Tosoian, J.J. Biomarkers for Detection of Clinically Significant Prostate Cancer: Contemporary Clinical Data and Future Directions. Transl. Androl. Urol. 2021, 10, 3091–3103. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.D.; Wender, R.C.; Etzioni, R.B.; Thompson, I.M.; D’Amico, A.V.; Volk, R.J.; Brooks, D.D.; Dash, C.; Guessous, I.; Andrews, K.; et al. American Cancer Society Guideline for the Early Detection of Prostate Cancer: Update 2010. CA Cancer J. Clin. 2010, 60, 70–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, I.M.; Pauler, D.K.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Parnes, H.L.; Minasian, L.M.; Ford, L.G.; Lippman, S.M.; Crawford, E.D.; et al. Prevalence of Prostate Cancer among Men with a Prostate-Specific Antigen Level ≤4.0 ng per Milliliter. N. Engl. J. Med. 2004, 350, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Catalona, W.J.; Partin, A.W.; Sanda, M.G.; Wei, J.T.; Klee, G.G.; Bangma, C.H.; Slawin, K.M.; Marks, L.S.; Loeb, S.; Broyles, D.L.; et al. A Multicenter Study of [-2]pro-prostate Specific Antigen Combined With Prostate Specific Antigen and Free Prostate Specific Antigen for Prostate Cancer Detection in the 2.0 to 10.0 ng/ml Prostate Specific Antigen Range. J. Urol. 2011, 185, 1650–1655. [Google Scholar] [CrossRef] [Green Version]
- Parekh, D.J.; Punnen, S.; Sjoberg, D.D.; Asroff, S.W.; Bailen, J.L.; Cochran, J.S.; Concepcion, R.; David, R.D.; Deck, K.B.; Dumbadze, I.; et al. A Multi-institutional Prospective Trial in the USA Confirms that the 4Kscore Accurately Identifies Men with High-grade Prostate Cancer. Eur. Urol. 2015, 68, 464–470. [Google Scholar] [CrossRef]
- Klocker, H.; Golding, B.; Weber, S.; Steiner, E.; Tennstedt, P.; Keller, T.; Schiess, R.; Gillessen, S.; Horninger, W.; Steuber, T. Development and Validation of a Novel Multivariate Risk Score to Guide Biopsy Decision for the Diagnosis of Clinically Significant Prostate Cancer. BJUI Compass 2020, 1, 15–20. [Google Scholar] [CrossRef]
- Shipitsin, M.; Small, C.; Choudhury, S.; Giladi, E.; Friedlander, S.; Nardone, J.; Hussain, S.; Hurley, A.D.; Ernst, C.; Huang, Y.E.; et al. Identification of Proteomic Biomarkers Predicting Prostate Cancer Aggressiveness and Lethality Despite Biopsy-sampling Error. Br. J. Cancer 2014, 111, 1201–1212. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Berman, D.M.; Rimm, D.L.; Shipitsin, M.; Putzi, M.; Nifong, T.P.; Small, C.; Choudhury, S.; Capela, T.; Coupal, L.; et al. Biology of Human Tumors Development and Clinical Validation of an In Situ Biopsy-based Multimarker Assay for Risk Stratification in Prostate Cancer. Clin. Cancer Res. 2015, 21, 2591–2600. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.L.; Anderson, N.G. The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Mol. Cell. Proteomics 2002, 1, 845–867. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.B.; Barrero, C.A.; Braverman, A.; Kim, P.D.; Jones, K.A.; Chen, D.E.; Bowler, R.P.; Merali, S.; Kelsen, S.G.; Yeung, A.T. Assessment of Two Immunodepletion Methods: Off-Target Effects and Variations in Immunodepletion Efficiency may Confound Plasma Proteomics. J. Proteome Res. 2012, 11, 5947–5958. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavoosidana, G.; Ronquist, G.; Darmanis, S.; Yan, J.; Carlsson, L.; Wu, D.; Conze, T.; Ek, P.; Semjonow, A.; Eltze, E.; et al. Multiple Recognition Assay Reveals Prostasomes as Promising Plasma Biomarkers for Prostate Cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 8809–8814. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Pang, B.; Zhu, Y.; Ni, J.; Thompson, J.; Malouf, D.; Bucci, J.; Graham, P.; Li, Y. Extracellular Vesicles: The Next Generation of Biomarkers for Liquid Biopsy-based Prostate Cancer Diagnosis. Theranostics 2020, 10, 2309–2326. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Timp, G. Beyond Mass Spectrometry, the Next Step in Proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Eng, J.K.; McCormack, A.L.; Schieltz, D. Method to Correlate Tandem Mass Spectra of Modified Peptides to Amino Acid Sequences in the Protein Database. Anal. Chem. 1995, 67, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Purvine, S.; Eppel, J.T.; Yi, E.C.; Goodlett, D.R. Shotgun Collision-induced Dissociation of Peptides Using a Time of Flight Mass Analyzer. Proteomics 2003, 3, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Venable, J.D.; Dong, M.Q.; Wohlschlegel, J.; Dillin, A.; Yates, J.R. Automated Approach for Quantitative Analysis of Complex Peptide Mixtures from Tandem Mass Spectra. Nat. Methods 2004, 1, 39–45. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [Green Version]
- Egertson, J.D.; Kuehn, A.; Merrihew, G.E.; Bateman, N.W.; MacLean, B.X.; Ting, Y.S.; Canterbury, J.D.; Marsh, D.M.; Kellmann, M.; Zabrouskov, V.; et al. Multiplexed MS/MS for Improved Data-independent Acquisition. Nat. Methods 2013, 10, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel Accumulation–serial Fragmentation Combined with Data-independent Acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem. 2016, 9, 1–24. [Google Scholar] [CrossRef]
- Wichmann, C.; Meier, F.; Winter, S.V.; Brunner, A.D.; Cox, J.; Mann, M. MaxQuant.live Enables Global Targeting of More Than 25,000 Peptides. Mol. Cell. Proteom. 2019, 18, 982–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Permentier, H.P.; Bischoff, R. Chemical Isotope Labeling for Quantitative Proteomics. Mass Spectrom. Rev. 2021, 1–31. [Google Scholar] [CrossRef]
- Thompson, A.; Wölmer, N.; Koncarevic, S.; Selzer, S.; Böhm, G.; Legner, H.; Schmid, P.; Kienle, S.; Penning, P.; Höhle, C.; et al. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-based Isobaric 16-plex Tandem Mass Tag Reagent Set. Anal. Chem. 2019, 91, 15941–15950. [Google Scholar] [CrossRef]
- Li, J.; Van Vranken, J.G.; Pontano Vaites, L.; Schweppe, D.K.; Huttlin, E.L.; Etienne, C.; Nandhikonda, P.; Viner, R.; Robitaille, A.M.; Thompson, A.H.; et al. TMTpro Reagents: A Set of Isobaric Labeling Mass Tags Enables Simultaneous Proteome-wide Measurements Across 16 Samples. Nat. Methods 2020, 17, 399–404. [Google Scholar] [CrossRef]
- Minogue, C.E.; Hebert, A.S.; Rensvold, J.W.; Westphall, M.S.; Pagliarini, D.J.; Coon, J.J. Multiplexed Quantification for Data-Independent Acquisition. Anal. Chem. 2015, 87, 2570–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, Y.; Zhang, Y.; Zhang, L.; Tao, T.; Lu, H. MdFDIA: A Mass Defect Based Four-Plex Data-Independent Acquisition Strategy for Proteome Quantification. Anal. Chem. 2017, 89, 10248–10255. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic Castration-resistant Prostate Cancer Reveals Intrapatient Similarity and Interpatient Heterogeneity of Therapeutic Kinase Targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, J.; Sethi, A.; Li, Q.K.; Chen, L.; Collins, B.; Gillet, L.C.J.; Wollscheid, B.; Zhang, H.; Aebersold, R. Glycoproteomic Analysis of Prostate Cancer Tissues by SWATH Mass Spectrometry Discovers N-acylethanolamine Acid Amidase and Protein Tyrosine Kinase 7 as Signatures for Tumor Aggressiveness. Mol. Cell. Proteom. 2014, 13, 1753–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; Lih, T.M.; Chen, S.-Y.; Cho, K.-C.; Eguez, R.V.; Höti, N.; Zhou, Y.; Yang, W.; Mangold, L.; Chan, D.W.; et al. Urinary Glycoproteins Associated with Aggressive Prostate Cancer. Theranostics 2020, 10, 11892–11907. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, W. Proteome-scale Analysis of Protein S-Acylation Comes of Age. J. Proteome Res. 2021, 20, 14–26. [Google Scholar] [CrossRef]
- Zhou, B.; An, M.; Freeman, M.R.; Yang, W. Technologies and Challenges in Proteomic Analysis of Protein S-acylation. J. Proteom. Bioinform. 2014, 7, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Di Vizio, D.; Kirchner, M.; Steen, H.; Freeman, M.R. Proteome-scale Characterization of Human S-acylated Proteins in Lipid Raft-enriched and Non-raft Membranes. Mol. Cell. Proteom. 2010, 9, 54–70. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wang, Y.; Yan, Y.; Mariscal, J.; Di Vizio, D.; Freeman, M.R.; Yang, W. Low-background Acyl-biotinyl Exchange Largely Eliminates the Coisolation of Non-S-acylated Proteins and Enables Deep S-acylproteomic Analysis. Anal. Chem. 2019, 91, 9858–9866. [Google Scholar] [CrossRef]
- Dowal, L.; Yang, W.; Freeman, M.R.; Steen, H.; Flaumenhaft, R. Proteomic Analysis of Palmitoylated Platelet Proteins. Blood 2011, 118, e62–e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariscal, J.; Vagner, T.; Kim, M.; Zhou, B.; Chin, A.; Zandian, M.; Freeman, M.R.; You, S.; Zijlstra, A.; Yang, W.; et al. Comprehensive Palmitoyl-Proteomic Analysis Identifies Distinct Protein Signatures for Large and Small Cancer-derived Extracellular Vesicles. J. Extracell. Vesicles 2020, 9, 1764192. [Google Scholar] [CrossRef]
- Sharma, C.; Yang, W.; Steen, H.; Freeman, M.R.; Hemler, M.E. Antioxidant Functions of DHHC3 Suppress Anti-cancer Drug Activities. Cell. Mol. Life Sci. 2021, 78, 2341–2353. [Google Scholar] [CrossRef]
- Cheng, S.S.; Yang, G.J.; Wang, W.; Leung, C.H.; Ma, D.L. The Design and Development of Covalent Protein-protein Interaction Inhibitors for Cancer Treatment. J. Hematol. Oncol. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Cheng, R.; Jackson, P.K. Identifying Cancer Drivers. Science 2021, 374, 38–39. [Google Scholar] [CrossRef]
- Zhou, B.; Yan, Y.; Wang, Y.; You, S.; Freeman, M.R.; Yang, W. Quantitative Proteomic Analysis of Prostate Tissue Specimens Identifies Deregulated Protein Complexes in Primary Prostate Cancer. Clin. Proteomics 2019, 16, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ablin, R.J.; Owen, S.; Jiang, W.G. Prostate Transglutaminase (TGase-4) Induces Epithelial-to-Mesenchymal Transition in Prostate Cancer Cells. Anticancer Res. 2017, 37, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Gato, D.; Wikström, P.; Tyanova, S.; Lavallee, C.; Thysell, E.; Carlsson, J.; Hägglöf, C.; Cox, J.; Andrén, O.; Stattin, P.; et al. The Proteome of Primary Prostate Cancer. Eur. Urol. 2016, 69, 942–952. [Google Scholar] [CrossRef]
- Myers, J.S.; von Lersner, A.K.; Sang, Q.X.A. Proteomic Upregulation of Fatty Acid Synthase and Fatty Acid Binding Protein 5 and Identification of Cancer- and Race-specific Pathway Associations in Human Prostate Cancer Tissues. J. Cancer 2016, 7, 1452–1464. [Google Scholar] [CrossRef] [Green Version]
- Staunton, L.; Tonry, C.; Lis, R.; Espina, V.; Liotta, L.; Inzitari, R.; Bowden, M.; Fabre, A.; O’Leary, J.; Finn, S.P.; et al. Pathology-driven Comprehensive Proteomic Profiling of the Prostate Cancer Tumor Microenvironment. Mol. Cancer Res. 2017, 15, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Li, L.; Zhong, Q.; Rupp, N.J.; Charmpi, K.; Wong, C.E.; Wagner, U.; Rueschoff, J.H.; Jochum, W.; Fankhauser, C.D.; et al. Multi-region Proteome Aanalysis Quantifies Spatial Heterogeneity of Prostate Tissue Biomarkers. Life Sci. Alliance 2018, 1, e201800042. [Google Scholar] [CrossRef] [Green Version]
- Latonen, L.; Afyounian, E.; Jylhä, A.; Nättinen, J.; Aapola, U.; Annala, M.; Kivinummi, K.K.; Tammela, T.T.L.; Beuerman, R.W.; Uusitalo, H.; et al. Integrative Proteomics in Prostate Cancer Uncovers Robustness Against Genomic and Transcriptomic Aberrations During Disease Progression. Nat. Commun. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.-K.; Föll, M.; Heckelmann, B.; Kiefer, S.; Werner, M.; Schilling, O.; Biniossek, M.L.; Jilg, C.A.; Drendel, V. Proteomic Characterization of Prostate Cancer to Distinguish Nonmetastasizing and Metastasizing Primary Tumors and Lymph Node Metastases. Neoplasia 2018, 20, 140–151. [Google Scholar] [CrossRef]
- Kawahara, R.; Recuero, S.; Nogueira, F.C.S.; Domont, G.B.; Leite, K.R.M.; Srougi, M.; Thaysen-Andersen, M.; Palmisano, G. Tissue Proteome Signatures Associated with Five Grades of Prostate Cancer and Benign Prostatic Hyperplasia. Proteomics 2019, 19. [Google Scholar] [CrossRef]
- Sinha, A.; Huang, V.; Livingstone, J.; Wang, J.; Fox, N.S.; Kurganovs, N.; Ignatchenko, V.; Fritsch, K.; Donmez, N.; Heisler, L.E.; et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 2019, 35, 414–427.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.K.; Ha, Y.S.; Na, A.Y.; Chun, S.Y.; Kwon, T.G.; Lee, J.N.; Lee, S. Identification of Novel Prognosis and Prediction Markers in Advanced Prostate Cancer Tissues Based on Quantitative Proteomics. Cancer Genom. Proteom. 2020, 17, 195–208. [Google Scholar] [CrossRef]
- Zhu, T.; Zhu, Y.; Xuan, Y.; Gao, H.; Cai, X.; Piersma, S.R.; Pham, T.V.; Schelfhorst, T.; Haas, R.R.G.D.; Bijnsdorp, I.V.; et al. DPHL: A DIA Pan-human Protein Mass Spectrometry Library for Robust Biomarker Discovery. Genom. Proteom. Bioinform. 2020, 18, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Latosinska, A.; Davalieva, K.; Makridakis, M.; Mullen, W.; Schanstra, J.P.; Vlahou, A.; Mischak, H.; Frantzi, M. Molecular Changes in Tissue Proteome during Prostate Cancer Development: Proof-of-Principle Investigation. Diagnostics 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ignatchenko, V.; Yao, C.Q.; Kalatskaya, I.; Nyalwidhe, J.O.; Lance, R.S.; Gramolini, A.O.; Troyer, D.A.; Stein, L.D.; Boutros, P.C.; et al. Identification of Differentially Expressed Proteins in Direct Expressed Prostatic Secretions of Men with Organ-confined Versus Extracapsular Prostate Cancer. Mol. Cell. Proteom. 2012, 11, 1870–1884. [Google Scholar] [CrossRef] [Green Version]
- Principe, S.; Kim, Y.; Fontana, S.; Ignatchenko, V.; Nyalwidhe, J.O.; Lance, R.S.; Troyer, D.A.; Alessandro, R.; Semmes, O.J.; Kislinger, T.; et al. Identification of Prostate-enriched Proteins by In-depth Proteomic Analyses of Expressed Prostatic Secretions in Urine. J. Proteome Res. 2012, 11, 2386–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Principe, S.; Jones, E.E.; Kim, Y.; Sinha, A.; Nyalwidhe, J.O.; Brooks, J.; Semmes, O.J.; Troyer, D.A.; Lance, R.S.; Kislinger, T.; et al. In-depth Proteomic Analyses of Exosomes Isolated from Expressed Prostatic Secretions in Urine. Proteomics 2013, 13, 1667–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K.; Kume, H.; Matsuzaki, K.; Kawashima, A.; Ujike, T.; Nagahara, A.; Uemura, M.; Miyagawa, Y.; Tomonaga, T.; Nonomura, N. Proteomic Analysis of Urinary Extracellular Vesicles from High Gleason Score Prostate Cancer. Sci. Rep. 2017, 7, 42961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhondt, B.; Geeurickx, E.; Tulkens, J.; Van Deun, J.; Vergauwen, G.; Lippens, L.; Miinalainen, I.; Rappu, P.; Heino, J.; Ost, P.; et al. Unravelling the Proteomic Landscape of Extracellular Vesicles in Prostate Cancer by Density-based Fractionation of Urine. J. Extracell. Vesicles 2020, 9, 1736935. [Google Scholar] [CrossRef] [PubMed]
- Jedinak, A.; Curatolo, A.; Zurakowski, D.; Dillon, S.; Bhasin, M.K.; Libermann, T.A.; Roy, R.; Sachdev, M.; Loughlin, K.R.; Moses, M.A. Novel Non-invasive Biomarkers that Distinguish Between Benign Prostate Hyperplasia and Prostate Cancer. BMC Cancer 2015, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Overbye, A.; Skotland, T.; Koehler, C.J.; Thiede, B.; Seierstad, T.; Berge, V.; Sandvig, K.; Llorente, A. Identification of Prostate Cancer Biomarkers in Urinary Exosomes. Oncotarget 2015, 6, 30357–30376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonry, C.L.; Doherty, D.; OShea, C.; Morrissey, B.; Staunton, L.; Flatley, B.; Shannon, A.; Armstrong, J.; Pennington, S.R. Discovery and Longitudinal Evaluation of Candidate Protein Biomarkers for Disease Recurrence in Prostate Cancer. J. Proteome Res. 2015, 14, 2769–2783. [Google Scholar] [CrossRef]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A.; Niu, L.; Müller, J.B.; Buchholtz, M.; Bader, J.M.; Teupser, D.; et al. Plasma Proteome Profiling to Detect and Avoid Sample-related Biases in Biomarker Studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Omenn, G.S.; Sun, Z.; Maes, M.; Pernemalm, M.; Palaniappan, K.K.; Letunica, N.; Vandenbrouck, Y.; Brun, V.; Tao, S.; et al. Advances and Utility of the Human Plasma Proteome. J. Proteome Res. 2021, 20, 5241–5263. [Google Scholar] [CrossRef]
- Alhamdani, M.S.; Schröder, C.; Hoheisel, J.D. Oncoproteomic Profiling with Antibody Microarrays. Genome Med. 2009, 1, 68. [Google Scholar] [CrossRef]
- Miller, J.C.; Zhou, H.; Kwekel, J.; Cavallo, R.; Burke, J.; Butler, E.B.; Teh, B.S.; Haab, B.B. Antibody Microarray Profiling of Human Prostate Cancer Sera: Antibody Screening and Identification of Potential Biomarkers. Proteomics 2003, 3, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Shafer, M.W.; Mangold, L.; Partin, A.W.; Haab, B.B. Antibody Array Profiling Reveals Serum TSP-1 as a Marker to Distinguish Benign from Malignant Prostatic Disease. Prostate 2007, 67, 255–267. [Google Scholar] [CrossRef]
- Heo, C.K.; Bahk, Y.Y.; Cho, E.W. Tumor-associated Autoantibodies as Diagnostic and Prognostic Biomarkers. BMB Rep. 2012, 45, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Adeola, H.A.; Smith, M.; Kaestner, L.; Blackburn, J.M.; Zerbini, L.F. Novel Potential Serological Prostate Cancer Biomarkers Using CT100+ Cancer Antigen Microarray Platform in a Multi-cultural South African cohort. Oncotarget 2016, 7, 13945–13964. [Google Scholar] [CrossRef]
- Lundberg, M.; Eriksson, A.; Tran, B.; Assarsson, E.; Fredriksson, S. Homogeneous Antibody-based Proximity Extension Assays Provide Sensitive and Specific Detection of Low-abundant Proteins in Human Blood. Nucleic Acids Res. 2011, 39, e102. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shen, M.; Hsu, E.-C.; Zhang, C.A.; Garcia-Marques, F.; Nolley, R.; Koul, K.; Rice, M.A.; Aslan, M.; Pitteri, S.J.; et al. Discovery of PTN as a Serum-based Biomarker of Pro-metastatic Prostate Cancer. Br. J. Cancer 2021, 124, 896–900. [Google Scholar] [CrossRef]
- Liu, S.; Garcia-Marques, F.; Zhang, C.A.; Lee, J.J.; Nolley, R.; Shen, M.; Hsu, E.C.; Aslan, M.; Koul, K.; Pitteri, S.J.; et al. Discovery of CASP8 as a Potential Biomarker for High-risk Prostate Cancer Through a High-multiplex Immunoassay. Sci. Rep. 2021, 11, 7612. [Google Scholar] [CrossRef] [PubMed]
- Campos-Fernández, E.; Oliveira Alqualo, N.; Moura Garcia, L.C.; Coutinho Horácio Alves, C.; Ferreira Arantes Vieira, T.D.; Caixeta Moreira, D.; Alonso-Goulart, V. The Use of Aptamers in Prostate Cancer: A Systematic Review of Theranostic Applications. Clin. Biochem. 2021, 93, 9–25. [Google Scholar] [CrossRef]
- Welton, J.L.; Brennan, P.; Gurney, M.; Webber, J.P.; Spary, L.K.; Carton, D.G.; Falcón-Pérez, J.M.; Walton, S.P.; Mason, M.D.; Tabi, Z.; et al. Proteomics Analysis of Vesicles Isolated From Plasma and Urine of Prostate Cancer Patients Using a Multiplex, Aptamer-based Protein Array. J. Extracell. Vesicles 2016, 5, 31209. [Google Scholar] [CrossRef] [Green Version]
- Dudani, J.S.; Ibrahim, M.; Kirkpatrick, J.; Warren, A.D.; Bhatia, S.N. Classification of Prostate Cancer Using a Protease Activity Nanosensor Library. Proc. Natl. Acad. Sci. USA 2018, 115, 8954–8959. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.A.; Chen, J.; Zhang, J.; Fornage, M.; Yang, Y.; Zhou, L.; Grams, M.E.; Tin, A.; Daya, N.; Hoogeveen, R.C.; et al. Large-scale Plasma Proteomic Analysis Identifies Proteins and Pathways Associated with Dementia Risk. Nat. Aging 2021, 1, 473–489. [Google Scholar] [CrossRef]
- Joshi, A.; Mayr, M. In Aptamers They Trust: Caveats of the SOMAscan Biomarker Discovery Platform from SomaLogic. Circulation 2018, 138, 2482–2485. [Google Scholar] [CrossRef]
- Tang, J.; Wang, Y.; Luo, Y.; Fu, J.; Zhang, Y.; Li, Y.; Xiao, Z.; Lou, Y.; Qiu, Y.; Zhu, F. Computational Advances of Tumor Marker Selection and Sample Classification in Cancer Proteomics. Comput. Struct. Biotechnol. J. 2020, 18, 2012–2025. [Google Scholar] [CrossRef] [PubMed]
- Clairefond, S.; Ouellet, V.; Péant, B.; Barrès, V.; Karakiewicz, P.I.; Mes-masson, A.M.; Saad, F. Expression of ERBB Family Members as Predictive Markers of Prostate Cancer Progression and Mortality. Cancers 2021, 13, 1688. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Naik, D.N.; Pothen, A.; Kasukurti, S.; Devineni, R.R.; Adam, B.L.; Semmes, O.J.; Wright, G.L. Computational Protein Biomarker Prediction: A Case Study for Prostate Cancer. BMC Bioinform. 2004, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Toth, R.; Schiffmann, H.; Hube-Magg, C.; Büscheck, F.; Höflmayer, D.; Weidemann, S.; Lebok, P.; Fraune, C.; Minner, S.; Schlomm, T.; et al. Random Forest-based Modelling to Detect Biomarkers for Prostate Cancer Progression. Clin. Epigenetics 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected Reaction Monitoring for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted Proteomic Quantification on Quadrupole-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [Green Version]
- Domon, B.; Gallien, S. Recent Advances in Targeted Proteomics for Clinical Applications. Proteom. Clin. Appl. 2015, 9, 423–431. [Google Scholar] [CrossRef]
- Faria, S.S.; Morris, C.F.M.; Silva, A.R.; Fonseca, M.P.; Forget, P.; Castro, M.S.; Fontes, W. A Timely Shift from Shotgun to Targeted Proteomics and How It Can Be Groundbreaking for Cancer Research. Front. Oncol. 2017, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Sequeiros, T.; Rigau, M.; Chiva, C.; Montes, M.; Garcia-Grau, I.; Garcia, M.; Diaz, S.; Celma, A.; Bijnsdorp, I.; Campos, A.; et al. Targeted Proteomics in Urinary Extracellular Vesicles Identifies Biomarkers for Diagnosis and Prognosis of Prostate Cancer. Oncotarget 2017, 8, 4960–4976. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.N.; Harlan, R.; Chen, J.; Aiyetan, P.; Liu, Y.; Sokoll, L.J.; Aebersold, R.; Chan, D.W.; Zhang, H. Multiplexed Targeted Mass Spectrometry-Based Assays for the Quantification of N-Linked Glycosite-Containing Peptides in Serum. Anal. Chem. 2015, 87, 10830–10838. [Google Scholar] [CrossRef] [Green Version]
- Erickson, B.K.; Rose, C.M.; Braun, C.R.; Erickson, A.R.; Knott, J.; McAlister, G.C.; Wühr, M.; Paulo, J.A.; Everley, R.A.; Gygi, S.P. A strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-throughput Protein Signature Characterization. Mol. Cell 2017, 65, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Garcia-D’Angeli, A.; Brennan, J.P.; Huo, Q. Predicting Detection Limits of Enzyme-linked Immunosorbent Assay (ELISA) and Bioanalytical Techniques in General. Analyst 2013, 139, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Whiteaker, J.R.; Paulovich, A.G. The Evolving Role of Mass Spectrometry in Cancer Biomarker Discovery. Cancer Biol. Ther. 2009, 8, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, A.H.; Diamandis, E.P.; Kulasingam, V. Uncovering the Depths of the Human Proteome: Antibody-based Technologies for Ultrasensitive Multiplexed Protein Detection and Quantification. Mol. Cell. Proteom. 2021, 100155. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Yamada, T. Signaling Pathway Profiling Using Reverse-phase Protein Array and Its Clinical Applications. Expert Rev. Proteom. 2017, 14, 607–615. [Google Scholar] [CrossRef]
- Akbani, R.; Becker, K.-F.; Carragher, N.; Goldstein, T.; de Koning, L.; Korf, U.; Liotta, L.; Mills, G.B.; Nishizuka, S.S.; Pawlak, M.; et al. Realizing the Promise of Reverse Phase Protein Arrays for Clinical, Translational, and Basic Research: A Workshop Report. Mol. Cell. Proteom. 2014, 13, 1625–1643. [Google Scholar] [CrossRef] [Green Version]
- Paweletz, C.P.; Charboneau, L.; Bichsel, V.E.; Simone, N.L.; Chen, T.; Gillespie, J.W.; Emmert-Buck, M.R.; Roth, M.J.; Petricoin III, E.F.; Liotta, L.A. Reverse Phase Protein Microarrays Which Capture Disease Progression Show Activation of Pro-survival Pathways at the Cancer Invasion Front. Oncogene 2001, 20, 1981–1989. [Google Scholar] [CrossRef] [Green Version]
- Grubb, R.L.; Calvert, V.S.; Wulkuhle, J.D.; Paweletz, C.P.; Linehan, W.M.; Phillips, J.L.; Chuaqui, R.; Valasco, A.; Gillespie, J.; Emmert-Buck, M.; et al. Signal Pathway Profiling of Prostate Cancer Using Reverse Phase Protein Arrays. Proteomics 2003, 3, 2142–2146. [Google Scholar] [CrossRef]
- Grubb, R.L.; Deng, J.; Pinto, P.A.; Mohler, J.L.; Chinnaiyan, A.; Rubin, M.; Linehan, W.M.; Liotta, L.A.; Petricoin, E.F.; Wulfkuhle, J.D. Pathway Biomarker Profiling of Localized and Metastatic Human Prostate Cancer Reveal Metastatic and Prognostic Signatures. J. Proteome Res. 2009, 8, 3044–3054. [Google Scholar] [CrossRef] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. Cancer Genome Atlas Research Network The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Pin, E.; Stratton, S.; Belluco, C.; Liotta, L.; Nagle, R.; Hodge, K.A.; Deng, J.; Dong, T.; Baldelli, E.; Petricoin, E.; et al. A Pilot Study Exploring the Molecular Architecture of the Tumor Microenvironment in Human Prostate Cancer Using Laser Capture Microdissection and Reverse Phase Protein Microarray. Mol. Oncol. 2016, 10, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Signore, M.; Alfonsi, R.; Federici, G.; Nanni, S.; Addario, A.; Bertuccini, L.; Aiello, A.; Di Pace, A.L.; Sperduti, I.; Muto, G.; et al. Diagnostic and Prognostic Potential of the Proteomic Profiling of Serum-Derived Extracellular Vesicles in Prostate Cancer. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kupcova Skalnikova, H.; Cizkova, J.; Cervenka, J.; Vodicka, P. Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. Int. J. Mol. Sci. 2017, 18, 2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaur, I.; Noack, A.; Makarevic, J.; Oppermann, E.; Waaga-Gasser, A.M.; Gasser, M.; Borgmann, H.; Huesch, T.; Gust, K.M.; Reiter, M.; et al. CCL2 Chemokine as a Potential Biomarker for Prostate Cancer: A Pilot Study. Cancer Res. Treat. 2015, 47, 306–312. [Google Scholar] [CrossRef]
- Al-Mazidi, S.; Farhat, K.; Nedjadi, T.; Chaudhary, A.; Zin Al-Abdin, O.; Rabah, D.; Al-Zoghaibi, M.; Djouhri, L. Association of Interleukin-6 and Other Cytokines With Self-reported Pain in Prostate Cancer Patients Receiving Chemotherapy. Pain Med. 2018, 19, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, N.D.; Pieczonka, C.M.; Henderson, R.J.; Bailen, J.L.; Saltzstein, D.R.; Concepcion, R.S.; Beebe-Dimmer, J.L.; Ruterbusch, J.J.; Levin, R.A.; Wissmueller, S.; et al. Development and Evaluation of the MiCheck Test for Aggressive Prostate Cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 683.e11–683.e18. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Biomarker | Sample Cohort | Source | Method | Ref. |
|---|---|---|---|---|
| NAAA, PTK7 | 10 normal, 24 non-aggressive PC, 16 aggressive PC, 25 metastatic PC | Tissue (OCT) | DIA-MS (label-free N-glycoproteomics) | [53] |
| MSK2, CPT2, COPA, NPY | 28 PC, 8 PC-adjacent normal | Tissue (FFPE) | DDA-MS (super-SILAC) | [66] |
| PGM3, PYCR1, GAA, HNRNPM, TALDO1, HNRNPL, GGCT, CTSH, NPEPPS, USP5, SUCLG2, HEXB, NDRG1, STEAP4, DDAH2, CTSD, COPA, TSTA3, PSMB5, TUFM, HSP90B1 | 14 PC, 9 matched non-malignant | Tissue (fresh frozen) | DDA-MS (label-free) | [67] |
| RPL28, RBM4, RPL5, NCL, ATP5H, THRAP3, H1FX, SNRPA1, RPL23, PPIB, TPD52, HNRNPL, HNRNPUL1, RALY, RPL10A, APEH, GOT1, USP14, RAB3D, DCXR, DPT, PPL, QDPR, SOD3, OLFML3, EPHX2, EMILIN1, FMOD, GDF15 | 4 GS3 + 3 PC, 4 GS4 + 4 PC | Tissue (frozen) | DDA-MS (label-free) | [68] |
| ATR, MRE11, RAD21, RAD23A, RAD23B, RAD50, RAD9A, CHEK1, XRCC5, XRCC6 | 12 BPH, 18 PC | Tissue (OCT) | DIA-MS (label-free) | [69] |
| ACO2, CS, FH, IDH3A, MDH2, OGDH, SUCLA2, SUCLG1 | 10 BPH, 17 untreated PC, 11 CRPC | Tissue (fresh frozen) | DIA-MS (label-free) | [70] |
| NDRG3, PARP1, ABHD11, SSH3 | 5 PC w/o metastasis, 5 PC w/lymph node metastases, 5 lymph node metastases | Tissue (FFPE) | DDA-MS (label-free) | [71] |
| IGKV3D-20, RNASET2, TACC2, ANXA7, LMOD1, PRCP, GYG1, NDUFV1, H1FX, APOBEC3C, CTSZ | 5 BPH, 50 PC | Tissue (fresh) | DDA-MS (label-free) | [72] |
| TGM2, NDRG3, KLK3/PSA, AKT1, PTEN, NKX3–1, KRAS, ATM | 76 PC | Tissue (OCT 1) | DDA-MS (label-free) | [73] |
| CARS2, NFKB2, ENPP4, PDSS2 (high-grade vs. low-grade); YBX1, SETSIP, FASN, PYCR1, PDSS2, FOLH1, SPON2 (high grade vs. normal); NSUN2, HEXB, HEXA, EPCAM, PYCR1 (low grade vs. normal) | 9 adjacent normal, 9 low-grade PC, 9 high-grade PC | Tissue (OCT) | DDA-MS (TMT) | [64] |
| SRM, NOLC1, PTGIS | 10 non-malignant, 8 PC, 2 metastatic | Tissue (frozen) | DDA-MS (TMT) | [74] |
| FASN, TPP1, SPON2 | 9 BPH, 8PC | Tissue | DIA-MS (label-free) | [75] |
| ALB, ACTG2, FLNA, MYH11, DES, TAGLN, COL6A3, HBB, ACTB, HIST1H2AH | 5 BPH, 17 PC | Tissue (fresh frozen) | DDA-MS (label-free) | [76] |
| SFN, MME, PARK7, TIMP1, TGM4 | 8 extracapsular, 8 organ-confined | Direct-EPS | DDA-MS (label-free) | [77] |
| KLK3/PSA, PAP, MSMB, FOLH1/PSMA, TMPRSS2 | 6 BPH, 5PC | EPS-urine | DDA-MS (label-free) | [78] |
| ACPP, ATRN, GP2, KLK11, PTPRN2, NPTN, CPE, RNASE2 (low in aggressive PC). CD97, ORM1, AFM, UMOD, PTGDS, GRN, SERPINA1, CLU, LRG1, LOX, DSC2 (high in aggressive PC) | 74 aggressive PC, 68 non-aggressive PC | EPS-urine | DIA-MS (label-free N-glycoproteomics) | [54] |
| KLK3/PSA, ACPP, TGM4, FOLH1/PSMA | 12 noncancer, 12 PC | EPS urinary EV | DDA-MS (label-free) | [79] |
| SCIN, AMBP, FABP5, CHMP4C, CHMP2B, BAIAP2, GRN, SYTL2, CALR, CHMP4A, DNPH1 | 11 negative biopsy, 18 PC including 5 GS6, 7 GS 7, and 6 GS 8–9 | EPS urinary EV | DDA-MS (iTRAQ 2) | [80] |
| KLK2, KLK3/PSA, FOLH1/PSMA, MSMB, ACPP, TGM4, NDRG1, NKX3-1, FKBP5, FAM129A, RAB27A, FASN, NEFH | 12 BPH, 12 PC | EPS urinary EV | DDA-MS (label-free) | [81] |
| B2M, PGA3, MUC3 | 83 BPH, 90 PC | Urine | DDA-MS (iTRAQ) | [82] |
| TM256/C17orf61, LAMPTOR1, VATL, ADIRF, KLK3/PSA, FOLH1/PSMA, TGM4, TMPRSS2, GOLPH3 | 15 noncancer, 17 PC | Urinary EV | DDA-MS (label-free) | [83] |
| C1QB, APOA4, CO9, ANT3, VTDB, PLMN, GPX3, ITIH4, CFAI, APOH, VTNC, IBP3, CLUS, APOA2, PEDF, TETN, CD14, LG3BP, CFAH, FCN3, HPT, CO3, APOA1, APOC3, SAMP, HEMO, CO6, KLK3/PSA, A2MG, A1At, APOE, A2Gl, TTHY, C1S, ZAG, AMBP, KNG1, CO4A, AACT, CAV1, TRFE | 3 PC with BCR, 3 control | Immunodepleted serum | DDA-MS (label-free) | [84] |
| Potential Biomarker | Sample Cohort | Source | Method | Ref. |
|---|---|---|---|---|
| FASN, TPP1, SPON2 | 16 BPH, 57 PC | Tissue | PRM-MS analysis of 6 peptides corresponding to 3 target proteins | [75] |
| ADSV, TGM4, CD63, GLPK5, SPHMPSA, PAPP | 54 noncancer, 22 low-grade PC, 31 high-grade PC | EPS urinary EV | SRM-MS analysis of 64 peptides corresponding to 64 target proteins | [109] |
| C1QB, APOA4, CO9, ANT3, VTDB, PLMN, GPX3, ITIH4, CFAI, APOH, VTNC, IBP3, CLUS, APOA2, PEDF, TETN, CD14, LG3BP, CFAH, FCN3, HPT, CO3, APOA1, APOC3, SAMP, HEMO, CO6, KLK3/PSA, A2MG, A1At, APOE, A2Gl, TTHY, C1S, ZAG, AMBP, KNG1, CO4A, AACT, CAV1, TRFE | 86 time-point samples from 3 PC patients with BCR and 3 controls | Immunodepleted serum | SRM-MS analysis of 59 peptides corresponding to 41 target proteins | [84] |
| ITIH2, CD44, IGHG2, CDH13 | 25 aggressive PC, 25 non-aggressive PC | Serum | PRM-MS analysis of 41 N-glycosite-containing peptides corresponding to 37 target proteins | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Yeon, S.Y.; Qian, C.; You, S.; Yang, W. On the Road to Accurate Protein Biomarkers in Prostate Cancer Diagnosis and Prognosis: Current Status and Future Advances. Int. J. Mol. Sci. 2021, 22, 13537. https://doi.org/10.3390/ijms222413537
Yan Y, Yeon SY, Qian C, You S, Yang W. On the Road to Accurate Protein Biomarkers in Prostate Cancer Diagnosis and Prognosis: Current Status and Future Advances. International Journal of Molecular Sciences. 2021; 22(24):13537. https://doi.org/10.3390/ijms222413537
Chicago/Turabian StyleYan, Yiwu, Su Yeon Yeon, Chen Qian, Sungyong You, and Wei Yang. 2021. "On the Road to Accurate Protein Biomarkers in Prostate Cancer Diagnosis and Prognosis: Current Status and Future Advances" International Journal of Molecular Sciences 22, no. 24: 13537. https://doi.org/10.3390/ijms222413537