
A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation

 , , ,
, , ,  , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
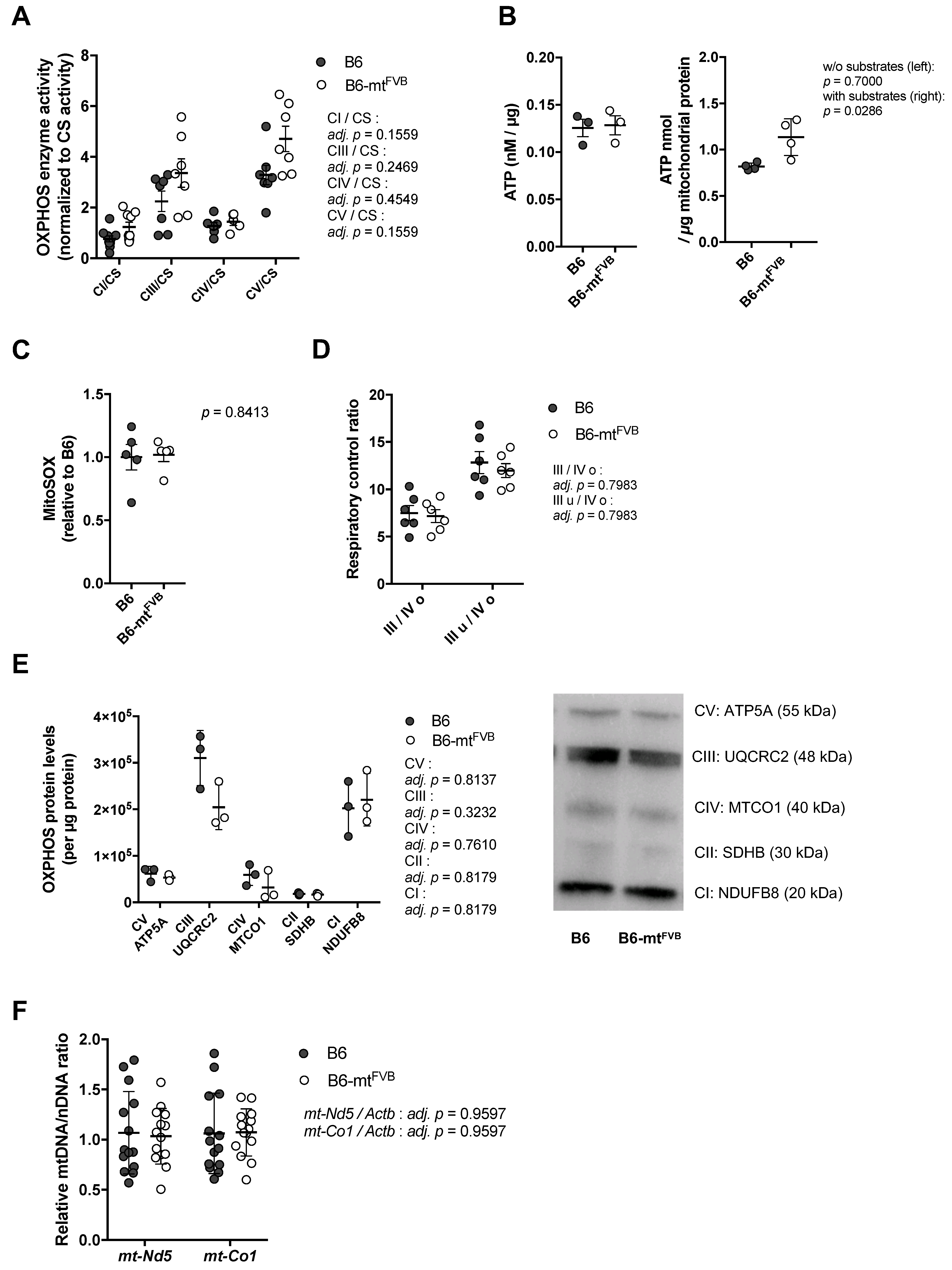
2.1. The Natural Polymorphism m.7778G > T in the mt-Atp8 Gene Influences Mitochondrial OXPHOS Function to a Moderate Extent
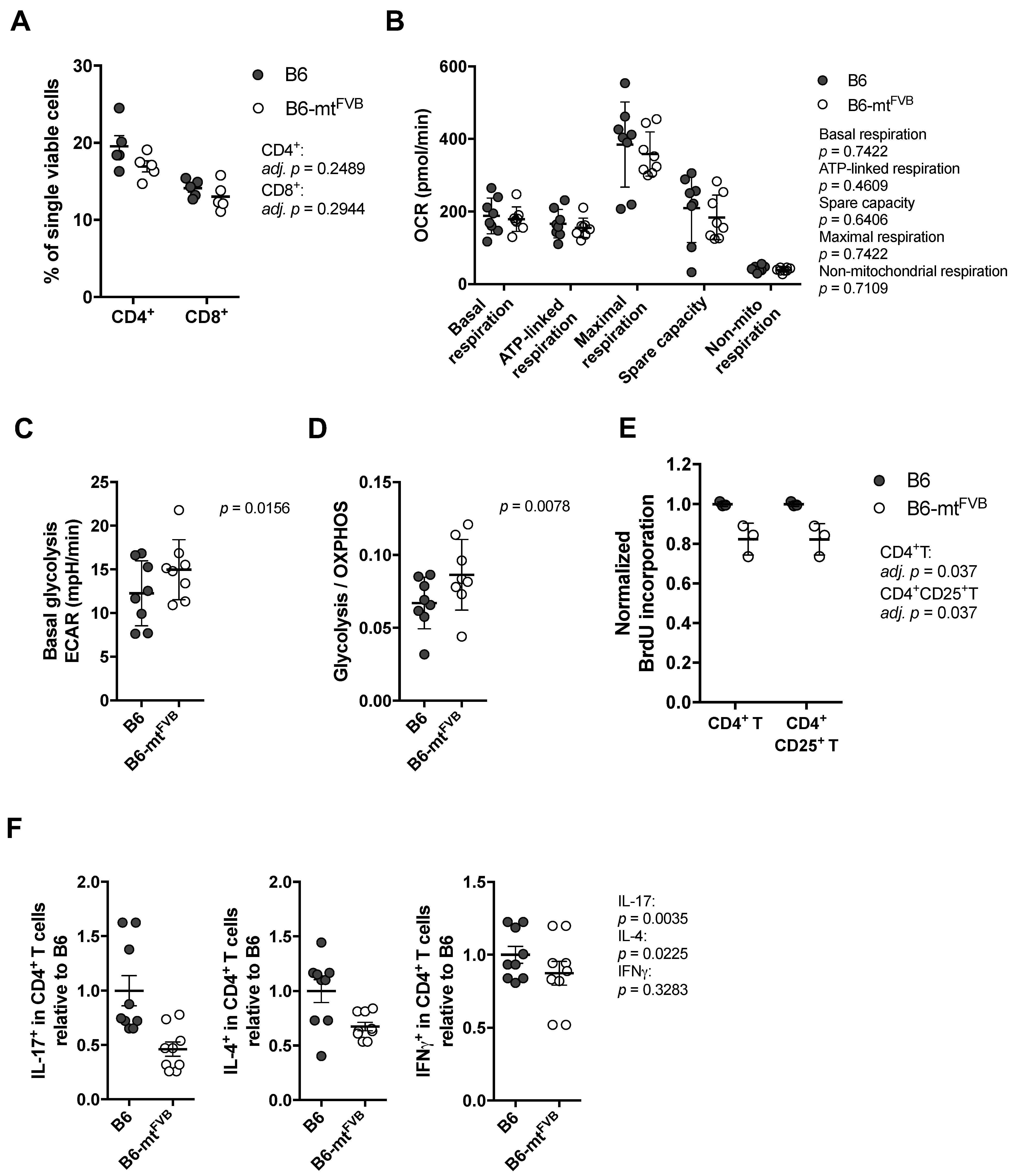
2.2. The Natural Variant m.7778G > T Results in a Differential Cellular Respiration Profile that Consequently Modified Cytokine Production in CD4+ T Cells
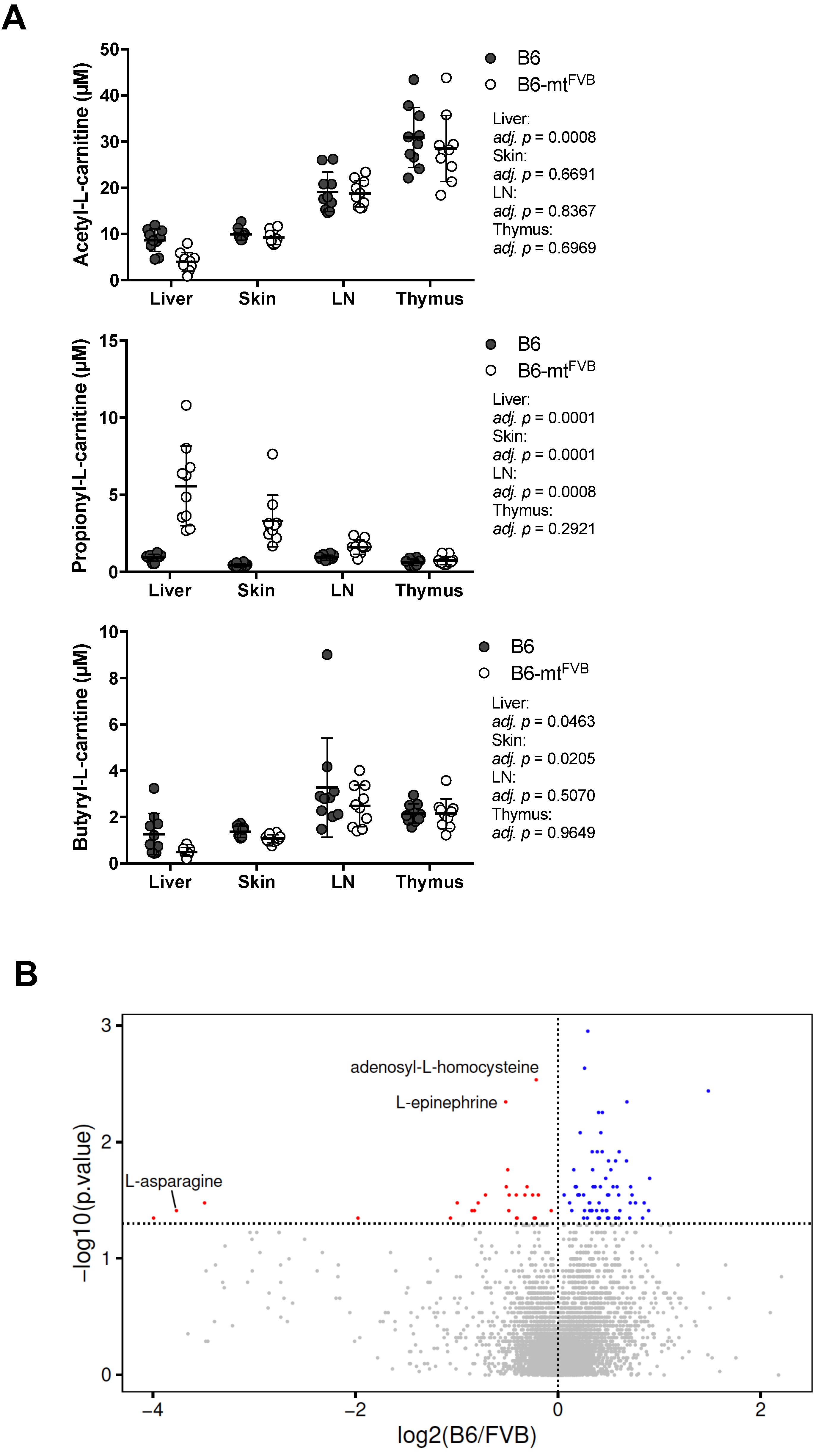
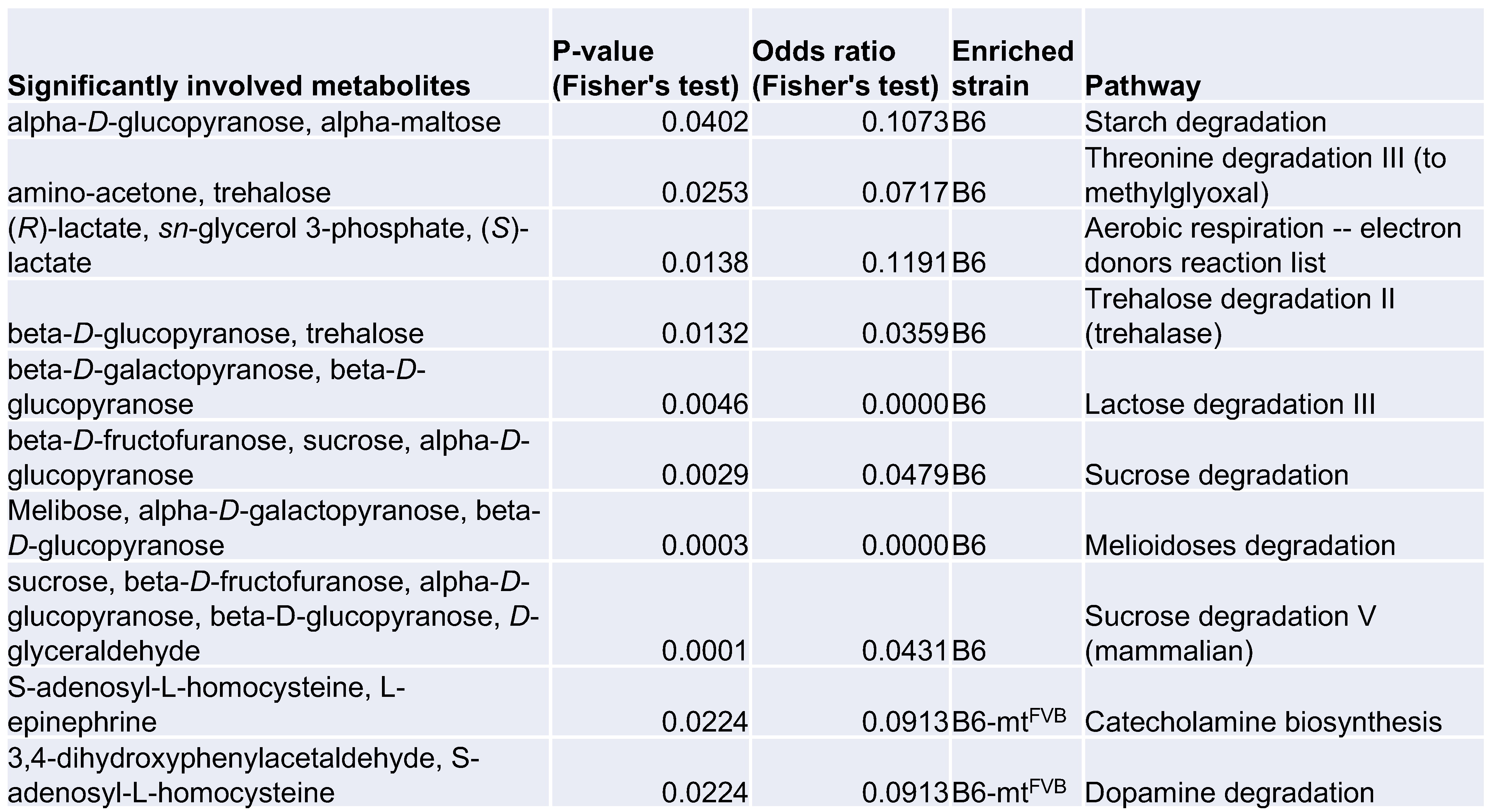
2.3. Differential Profile of Metabolites, Including Short Chain Fatty Acids, in Mice Carrying a Mutation in the mt-Atp8 Gene
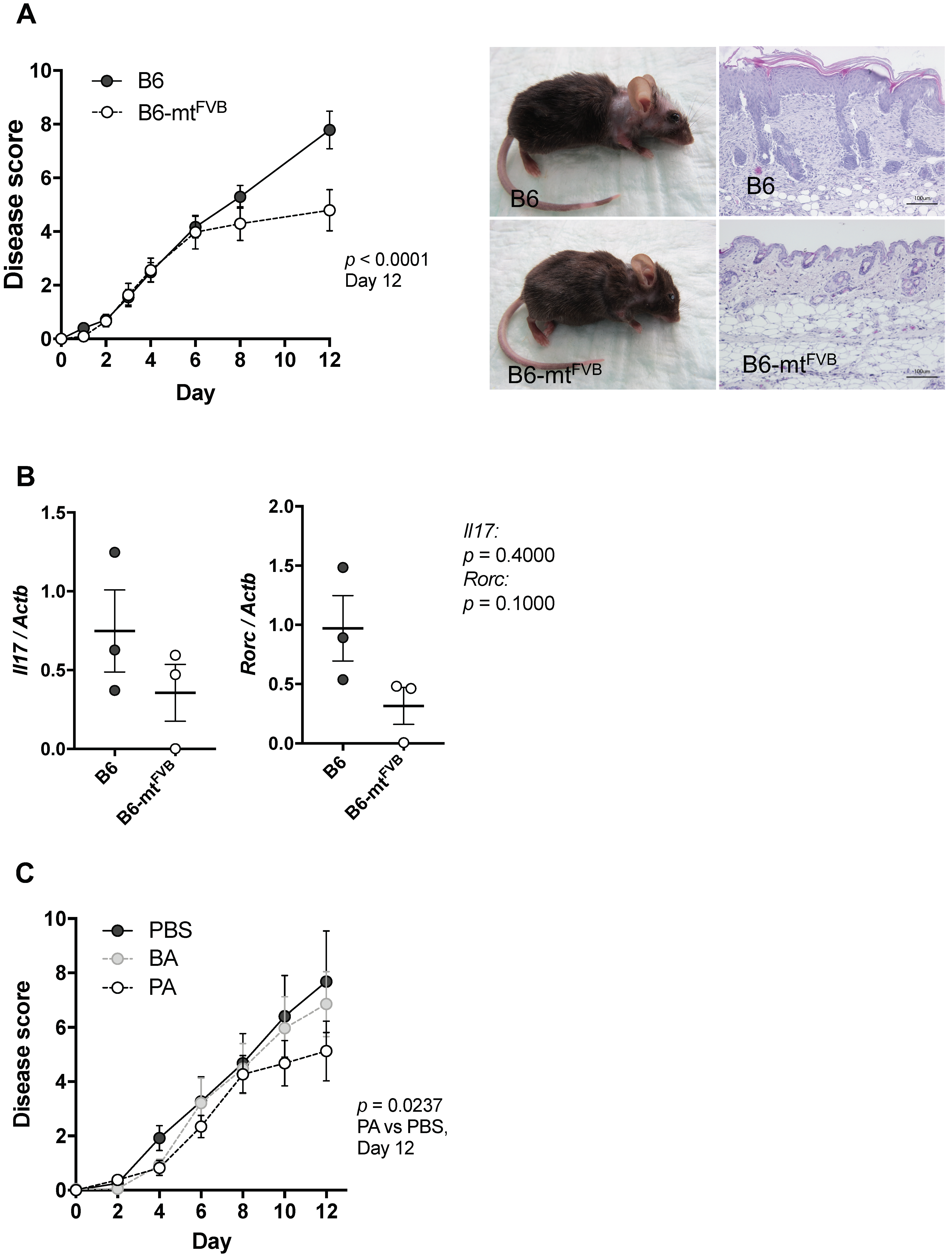
2.4. The mt-Atp8 Variant (m.7778G > T) Results in Milder Experimental Skin Inflammation than the Wild-Type Allele, a Phenomenon Mimicked by Propionate Treatment
3. Discussion
4. Materials and Methods
4.1. Mouse Husbandry
4.2. Isolation of Liver Mitochondria
4.3. Determination of ATP and Mitochondrial ROS Levels in Isolated Mitochondria
4.4. OXPHOS Enzyme Activity Measurement
4.5. Western Blot Analysis
4.6. mtDNA Copy Number
4.7. Immune Cell Preparation
4.8. Flow Cytometry Analysis
4.9. Mitochondrial Superoxide and Mitochondrial Membrane Potential in CD4+T Cells
4.10. ATP Content in CD4+ T Cells
4.11. Cellular Metabolism Flux Assay Using Seahorse Bioanalyzer
4.12. Neutrophil Granulocyte Functional Phenotyping
4.13. Untargeted Metabolomics
4.14. An Integrated Analysis of Untargeted Metabolomics Data and Gene Array Expression Data
4.15. Autoantibody-Transfer Experimental Autoimmune Skin Inflammation (Epidermolysis Bullosa Acquisita; EBA) Model
4.16. Imiquimod (Aldara®)-Induced Psoriasisform Dermatitis (AIPD) Model
4.17. Histopathology
4.18. Quantitative Real-Time PCR
4.19. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, D.C.; Zheng, X.X.; Lott, M.T.; Shoffner, J.M.; Hodge, J.A.; Kelley, R.I.; Epstein, C.M.; Hopkins, L.C. Familial Mitochondrial Encephalomyopathy (MERRF): Genetic, Pathophysiological, and Biochemical Characterization of a Mitochondrial DNA Disease. Cell 1988, 55, 601–610. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Starikovskaya, E.; Derbeneva, O.; Hosseini, S.; Allen, J.C.; Mikhailovskaya, I.E.; Sukernik, R.I.; Wallace, D.C. The Role of MtDNA Background in Disease Expression: A New Primary LHON Mutation Associated with Western Eurasian Haplogroup. J. Hum. Genet. 2002, 110, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse MtDNA Mutant Model of Leber Hereditary Optic Neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef]
- Hashizume, O.; Shimizu, A.; Yokota, M.; Sugiyama, A.; Nakada, K.; Miyoshi, H.; Itami, M.; Ohira, M.; Nagase, H.; Takenaga, K.; et al. Specific Mitochondrial DNA Mutation in Mice Regulates Diabetes and Lymphoma Development. Proc. Natl. Acad. Sci. USA 2012, 109, 10528–10533. [Google Scholar] [CrossRef]
- Hirose, M.; Schilf, P.; Gupta, Y.; Wright, M.N.; Wright, M.N.; Jöhren, O.; Wagner, A.E.; Sina, C.; Ziegler, A.; Ristow, M.; et al. Lifespan Effects of Mitochondrial Mutations. Nature 2016, 540, E13–E14. [Google Scholar] [CrossRef]
- Hirose, M.; Schilf, P.; Gupta, Y.; Zarse, K.; Künstner, A.; Fähnrich, A.; Busch, H.; Yin, J.; Wright, M.N.; Ziegler, A.; et al. Low-Level Mitochondrial Heteroplasmy Modulates DNA Replication, Glucose Metabolism and Lifespan in Mice. Sci. Rep. 2018, 8, 5872. [Google Scholar] [CrossRef]
- Hirose, M.; Künstner, A.; Schilf, P.; Tietjen, A.K.; Jöhren, O.; Huebbe, P.; Rimbach, G.; Rupp, J.; Schwaninger, M.; Busch, H.; et al. A Natural MtDNA Polymorphism in Complex III Is a Modifier of Healthspan in Mice. Int. J. Mol. Sci. 2019, 20, 2359. [Google Scholar] [CrossRef]
- Hirose, M.; Schilf, P.; Zarse, K.; Busch, H.; Fuellen, G.; Jöhren, O.; Köhling, R.; König, I.R.; Richer, B.; Rupp, J.; et al. Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan. Genes 2019, 10, 532. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and Nuclear DNA Matching Shapes Metabolism and Healthy Ageing. Nature 2016. [Google Scholar] [CrossRef]
- Galati, D.; Srinivasan, S.; Raza, H.; Prabu, S.K.; Hardy, M.; Chandran, K.; Lopez, M.; Kalyanaraman, B.; Avadhani, N.G. Role of Nuclear-Encoded Subunit Vb in the Assembly and Stability of Cytochrome c Oxidase Complex: Implications in Mitochondrial Dysfunction and ROS Production. Biochem. J. 2009, 420, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The Mitochondrial Respiratory Chain Is Essential for Haematopoietic Stem Cell Function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Loveland, B.; Wang, C.R.; Yonekawa, H.; Hermel, E.; Lindahl, K.F. Maternally Transmitted Histocompatibility Antigen of Mice: A Hydrophobic Peptide of a Mitochondrially Encoded Protein. Cell 1990, 60, 971–980. [Google Scholar] [CrossRef]
- Ishikawa, K.; Toyama-Sorimachi, N.; Nakada, K.; Morimoto, M.; Imanishi, H.; Yoshizaki, M.; Sasawatari, S.; Niikura, M.; Takenaga, K.; Yonekawa, H.; et al. The Innate Immune System in Host Mice Targets Cells with Allogenic Mitochondrial DNA. J. Exp. Med. 2010, 207, 2297–2305. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397. [Google Scholar] [CrossRef]
- O’Sullivan, D.; van der Windt, G.J.W.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef]
- van der Windt, G.J.W.; Everts, B.; Chang, C.-H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bähre, H.; et al. De Novo Fatty Acid Synthesis Controls the Fate between Regulatory T and T Helper 17 Cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like Receptor Signaling Balance Treg Cell Anabolic Metabolism for Suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Priyadharshini, B.; Loschi, M.; Newton, R.H.; Zhang, J.-W.; Finn, K.K.; Gerriets, V.A.; Huynh, A.; Rathmell, J.C.; Blazar, B.R.; Turka, L.A. Cutting Edge: TGF-β and Phosphatidylinositol 3-Kinase Signals Modulate Distinct Metabolism of Regulatory T Cell Subsets. J. Immunol. 2018, 201, 2215–2219. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate Is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef]
- Mehta, M.M.; Weinberg, S.E.; Chandel, N.S. Mitochondrial Control of Immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608–620. [Google Scholar] [CrossRef]
- Sack, M.N. Mitochondrial Fidelity and Metabolic Agility Control Immune Cell Fate and Function. J. Clin. Investig. 2018, 128, 3651–3661. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Acín-Pérez, R.; Mullikin, J.C.; Park, J.S.; Moreno-Loshuertos, R.; Hu, P.; Pérez-Martos, A.; Fernández-Silva, P.; Bai, Y.; Enríquez, J.A. Revisiting the Mouse Mitochondrial DNA Sequence. Nucleic Acids Res. 2003, 31, 5349–5355. [Google Scholar] [CrossRef]
- Ferris, S.D.; Sage, R.D.; Prager, E.M.; Ritte, U.; Wilson, A.C. Mitochondrial DNA Evolution in Mice. Genetics 1983, 105, 681–721. [Google Scholar] [CrossRef] [PubMed]
- Goios, A.; Pereira, L.; Bogue, M.; Macaulay, V.; Amorim, A. MtDNA Phylogeny and Evolution of Laboratory Mouse Strains. Genome Res. 2007, 17, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Gimsa, U.; Wester-Rosenlöf, L.; Kanitz, E.; Otten, W.; Kunz, M.; Ibrahim, S.M. Dissecting the Effects of MtDNA Variations on Complex Traits Using Mouse Conplastic Strains. Genome Res. 2009, 19, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sitaru, C.; Mihai, S.; Otto, C.; Chiriac, M.T.; Hausser, I.; Dotterweich, B.; Saito, H.; Rose, C.; Ishiko, A.; Zillikens, D. Induction of Dermal-Epidermal Separation in Mice by Passive Transfer of Antibodies Specific to Type VII Collagen. J. Clin. Investig. 2005, 115, 870–878. [Google Scholar] [CrossRef] [PubMed]
- van der Fits, L.; Mourits, S.; Voerman, J.S.A.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.-M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-Induced Psoriasis-like Skin Inflammation in Mice Is Mediated via the IL-23/IL-17 Axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef] [PubMed]
- Schröder, T.; Kucharczyk, D.; Bär, F.; Pagel, R.; Derer, S.; Jendrek, S.T.; Sünderhauf, A.; Brethack, A.-K.; Hirose, M.; Möller, S.; et al. Mitochondrial Gene Polymorphisms Alter Hepatic Cellular Energy Metabolism and Aggravate Diet-Induced Non-Alcoholic Steatohepatitis. Mol. Metab. 2016, 5, 283–295. [Google Scholar] [CrossRef]
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16. [Google Scholar] [CrossRef]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.-H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial Genetic Medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic Epilepsy and Ragged-Red Fiber Disease (MERRF) Is Associated with a Mitochondrial DNA TRNA(Lys) Mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T Cell Metabolism Drives Immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, Y.; Hao, L.-Y.; Liu, X.; Lesch, C.; Sanchez, B.M.; Wendling, J.M.; Morgan, R.W.; Aicher, T.D.; Carter, L.L.; et al. Sterol Metabolism Controls TH17 Differentiation by Generating Endogenous RORγ Agonists. Nat. Chem. Biol. 2015, 11, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Hellman, J.; Horswill, A.R.; Crosby, H.A.; Francis, K.P.; Prakash, A. Elevated Gut Microbiome-Derived Propionate Levels Are Associated With Reduced Sterile Lung Inflammation and Bacterial Immunity in Mice. Front. Microbiol. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Künstner, A.; Schilf, P.; Sünderhauf, A.; Rupp, J.; Jöhren, O.; Schwaninger, M.; Sina, C.; Baines, J.F.; Ibrahim, S.M. Mitochondrial Gene Polymorphism Is Associated with Gut Microbial Communities in Mice. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; McWilliam Leitch, C.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic Distribution of Three Pathways for Propionate Production within the Human Gut Microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef]
- Siliprandi, N.; Di Lisa, F.; Menabò, R. Propionyl-L-Carnitine: Biochemical Significance and Possible Role in Cardiac Metabolism. Cardiovasc. Drugs Ther. 1991, 5, 11–15. [Google Scholar] [CrossRef]
- Chakievska, L.; Holtsche, M.M.; Künstner, A.; Goletz, S.; Petersen, B.-S.; Thaci, D.; Ibrahim, S.M.; Ludwig, R.J.; Franke, A.; Sadik, C.D.; et al. IL-17A Is Functionally Relevant and a Potential Therapeutic Target in Bullous Pemphigoid. J. Autoimmun. 2019, 96, 104–112. [Google Scholar] [CrossRef]
- Boutet, M.-A.; Nerviani, A.; Gallo Afflitto, G.; Pitzalis, C. Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints. Int. J. Mol. Sci. 2018, 19, 530. [Google Scholar] [CrossRef]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of Immune Cell Function by Short-Chain Fatty Acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Perry, R.J.; Borders, C.B.; Cline, G.W.; Zhang, X.M.; Alves, T.C.; Petersen, K.F.; Rothman, D.L.; Kibbey, R.G.; Shulman, G.I. Propionate Increases Hepatic Pyruvate Cycling and Anaplerosis and Alters Mitochondrial Metabolism. J. Biol. Chem. 2016, 291, 12161–12170. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Kotani, J.; Usami, M. Butyrate and Propionate Induced Activated or Non-Activated Neutrophil Apoptosis via HDAC Inhibitor Activity but without Activating GPR-41/GPR-43 Pathways. Nutrition 2010, 26, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut Microbiota Metabolism of Dietary Fiber Influences Allergic Airway Disease and Hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Boominathan, A.; Vanhoozer, S.; Basisty, N.; Powers, K.; Crampton, A.L.; Wang, X.; Friedricks, N.; Schilling, B.; Brand, M.D.; O’Connor, M.S. Stable Nuclear Expression of ATP8 and ATP6 Genes Rescues a MtDNA Complex V Null Mutant. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef]
- Bornhövd, C.; Vogel, F.; Neupert, W.; Reichert, A.S. Mitochondrial Membrane Potential Is Dependent on the Oligomeric State of F1F0-ATP Synthase Supracomplexes. J. Biol. Chem. 2006, 281, 13990–13998. [Google Scholar] [CrossRef]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gomez, J.D.; Kuhlbrandt, W. Structure of the Yeast F1Fo-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef]
- Wittig, I.; Meyer, B.; Heide, H.; Steger, M.; Bleier, L.; Wumaier, Z.; Karas, M.; Schägger, H. Assembly and Oligomerization of Human ATP Synthase Lacking Mitochondrial Subunits a and A6L. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1004–1011. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the Dark Side of Glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- Nokin, M.-J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic Potential of Methylglyoxal, a Side-Product of Glycolysis, in Switching Tumours from Growth to Death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of MtDNA/NDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Johnson, M.S.; Hardy, R.W.; Ballinger, S.W.; Darley-Usmar, V.M. Methods for Defining Distinct Bioenergetic Profiles in Platelets, Lymphocytes, Monocytes, and Neutrophils, and the Oxidative Burst from Human Blood. Lab. Investig. 2013, 93, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A Review of the Mitochondrial and Glycolytic Metabolism in Human Platelets and Leukocytes: Implications for Their Use as Bioenergetic Biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.W.; Brand, M.D.; Petrosyan, S.; Ashok, D.; Elorza, A.A.; Ferrick, D.A.; Murphy, A.N. High Throughput Microplate Respiratory Measurements Using Minimal Quantities of Isolated Mitochondria. PLoS ONE 2011, 6, e0021746. [Google Scholar] [CrossRef]
- Iwata, H.; Witte, M.; Samavedam, U.K.S.R.L.; Gupta, Y.; Shimizu, A.; Ishiko, A.; Schroder, T.; Seeger, K.; Dahlke, M.; Rades, D.; et al. Radiosensitive Hematopoietic Cells Determine the Extent of Skin Inflammation in Experimental Epidermolysis Bullosa Acquisita. J. Immunol. 2015, 195, 1945–1954. [Google Scholar] [CrossRef]
- Evsikov, A.V.; Dolan, M.E.; Genrich, M.P.; Patek, E.; Bult, C.J. MouseCyc: A Curated Biochemical Pathways Database for the Laboratory Mouse. Genome Biol. 2009, 10, R84. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes and the BioCyc Collection of Pathway/Genome Databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Sezin, T.; Zillikens, D.; Sadik, C. Leukotrienes Do Not Modulate the Course of AldaraTM-Induced Psoriasiform Dermatitis in Mice. Acta Derm. Venereol. 2015, 95, 341–342. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilf, P.; Künstner, A.; Olbrich, M.; Waschina, S.; Fuchs, B.; Galuska, C.E.; Braun, A.; Neuschütz, K.; Seutter, M.; Bieber, K.; et al. A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation. Int. J. Mol. Sci. 2021, 22, 1006. https://doi.org/10.3390/ijms22031006
Schilf P, Künstner A, Olbrich M, Waschina S, Fuchs B, Galuska CE, Braun A, Neuschütz K, Seutter M, Bieber K, et al. A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation. International Journal of Molecular Sciences. 2021; 22(3):1006. https://doi.org/10.3390/ijms22031006
Chicago/Turabian StyleSchilf, Paul, Axel Künstner, Michael Olbrich, Silvio Waschina, Beate Fuchs, Christina E. Galuska, Anne Braun, Kerstin Neuschütz, Malte Seutter, Katja Bieber, and et al. 2021. "A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation" International Journal of Molecular Sciences 22, no. 3: 1006. https://doi.org/10.3390/ijms22031006
APA StyleSchilf, P., Künstner, A., Olbrich, M., Waschina, S., Fuchs, B., Galuska, C. E., Braun, A., Neuschütz, K., Seutter, M., Bieber, K., Hellberg, L., Sina, C., Laskay, T., Rupp, J., Ludwig, R. J., Zillikens, D., Busch, H., Sadik, C. D., Hirose, M., & Ibrahim, S. M. (2021). A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation. International Journal of Molecular Sciences, 22(3), 1006. https://doi.org/10.3390/ijms22031006