
Glucocorticoid Receptor Regulates TNFSF11 Transcription by Binding to Glucocorticoid Responsive Element in TNFSF11 Proximal Promoter Region

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
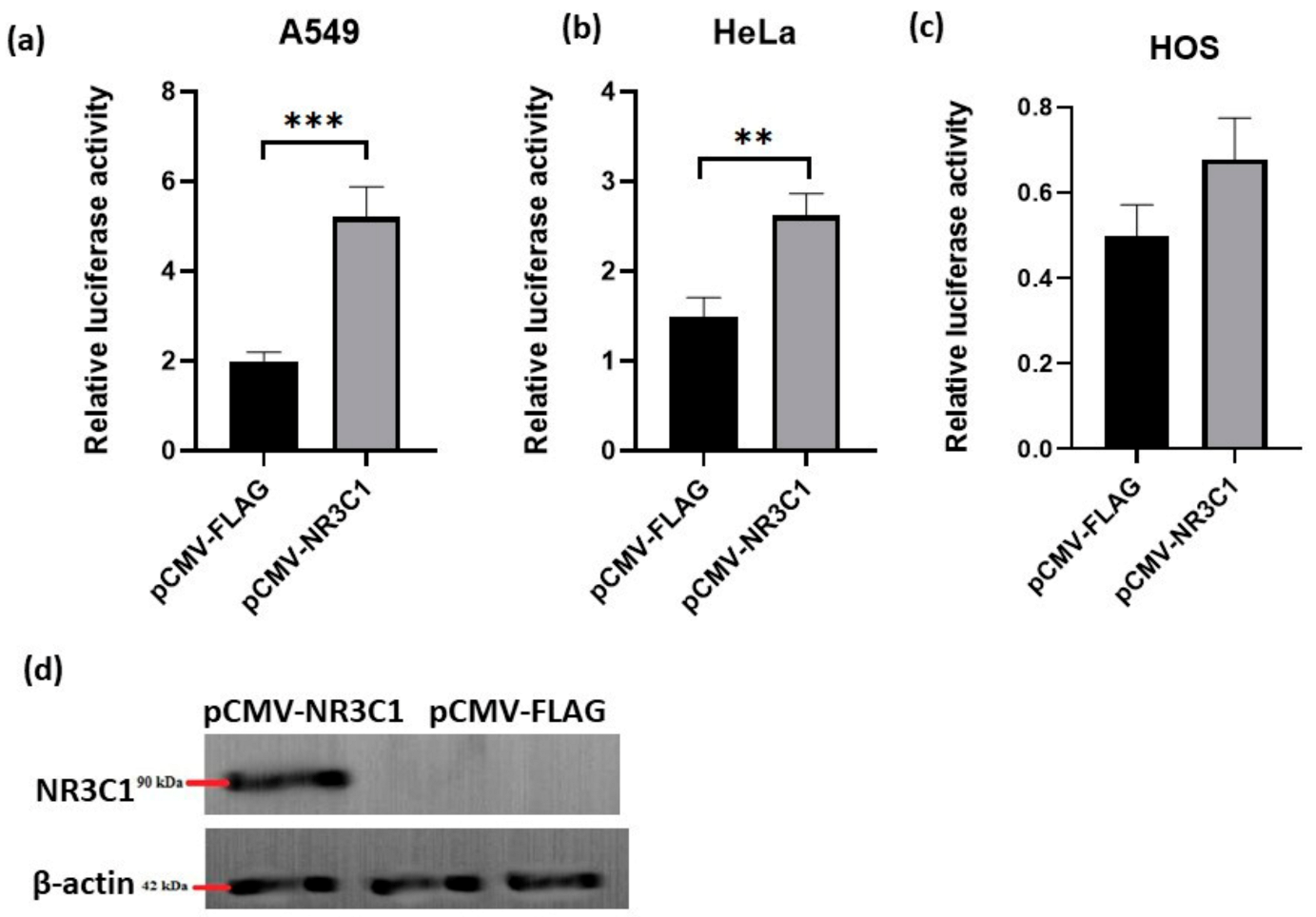
2.1. Overexpressed Glucocorticoid Receptor NR3C1 Increases RANKL Promoter Activity in A549, HOS and HeLa Cells
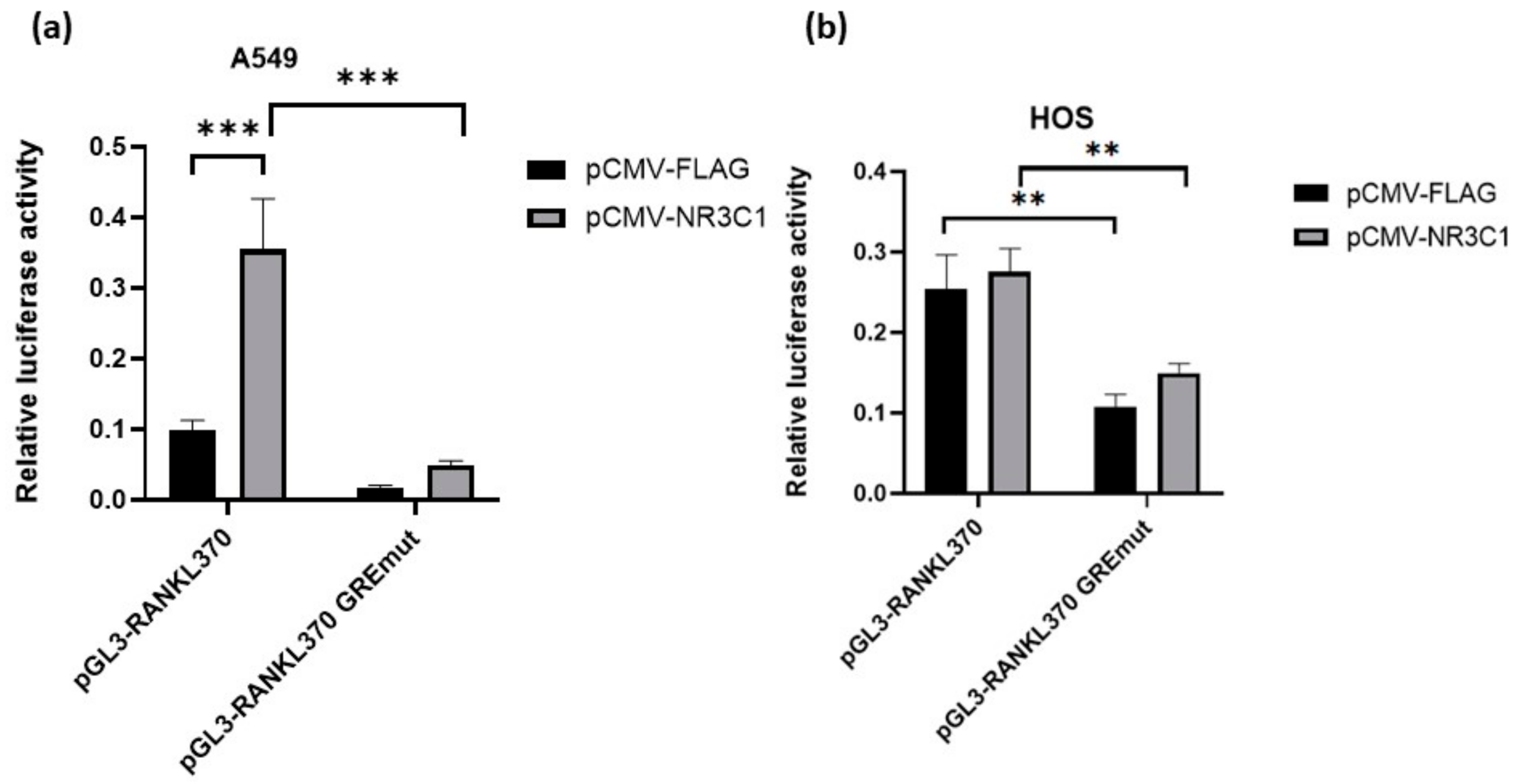
2.2. Mutation in Glucocorticoid Responsive Element Alleviates Effect of Glucocorticoid Receptor on RANKL Promoter Activity
2.3. Glucocorticoid Receptor Binds to the RANKL Proximal Promoter Region
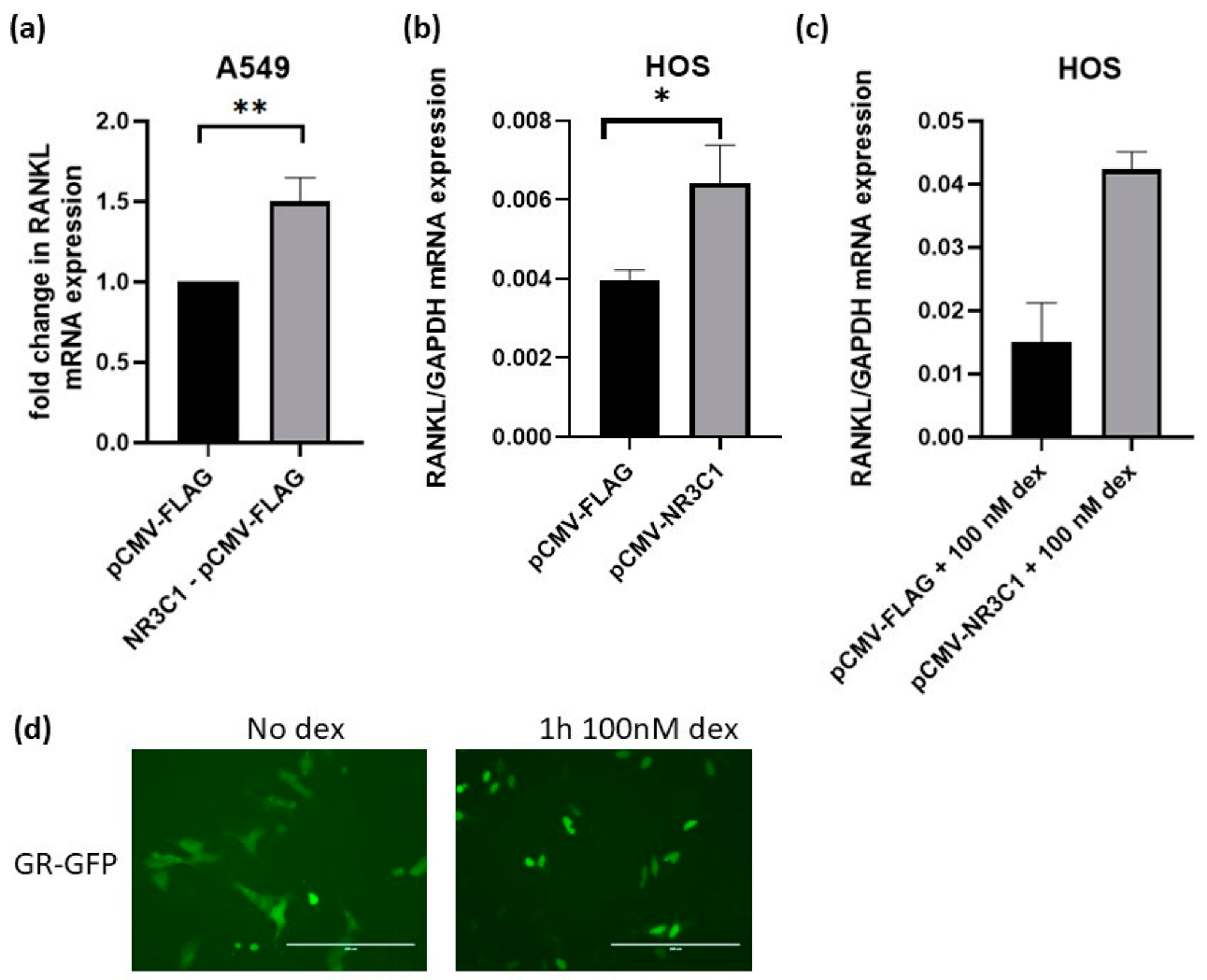
2.4. Overexpression of Glucocorticoid Receptor Upregulates RANKL Expression in A549 and HOS Cells
3. Discussion
4. Materials and Methods
4.1. Plasmid Constructs
4.2. Cell Culturing and Transfections
4.3. Luciferase Reporter Assay
4.4. Quantitative PCR
4.5. Western Blotting
4.6. RNA Interference
4.7. Electrophoretic Mobility Shift Assays
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RANKL | receptor activator of nuclear factor κ B ligand |
| GIO | Glucocorticoid induced osteoporosis |
| GR | Glucocorticoid receptor |
| GRE | glucocorticoid responsive element |
| GC | glucocorticoid |
| NR3C1 | Nuclear Receptor Subfamily 3 Group C Member |
| OPG | osteoprotegerin |
| EMSA | electrophoretic mobility shift assay |
References
- Moutsatsou, P.; Kassi, E.; Papavassiliou, A.G. Glucocorticoid receptor signaling in bone cells. Trends Mol. Med. 2012, 18, 348–359. [Google Scholar] [CrossRef]
- Chotiyarnwong, P.; McCloskey, E.V. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat. Rev. Endocrinol. 2020, 16, 437–447. [Google Scholar] [CrossRef]
- Compston, J. Glucocorticoid-induced osteoporosis: An update. Endocrine 2018, 61, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J.P. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef]
- Ahmad, M.; Hachemi, Y.; Paxian, K.; Mengele, F.; Koenen, M.; Tuckermann, J. A Jack of All Trades: Impact of Glucocorticoids on Cellular Cross-Talk in Osteoimmunology. Front. Immunol. 2019, 10, 2460. [Google Scholar] [CrossRef] [Green Version]
- Adami, G.; Saag, K.G. Glucocorticoid-induced osteoporosis update. Curr. Opin. Rheumatol. 2019, 31, 388–393. [Google Scholar] [CrossRef]
- Swanson, C.; Lorentzon, M.; Conaway, H.H.; Lerner, U.H. Glucocorticoid regulation of osteoclast differentiation and expression of receptor activator of nuclear factor-kappaB (NF-kappaB) ligand, osteoprotegerin, and receptor activator of NF-kappaB in mouse calvarial bones. Endocrinology 2006, 147, 3613–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Gori, F.; Riggs, B.L.; Lacey, D.L.; Dunstan, C.R.; Spelsberg, T.C.; Khosla, S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: Potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 1999, 140, 4382–4389. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.Y.; Peacock, M.; Bellido, T. Glucocorticoid Excess in Bone and Muscle. Clin. Rev. Bone Miner. Metab. 2018, 16, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, I.; Merlotti, D.; Falchetti, A.; Gennari, L. Treatment options for glucocorticoid-induced osteoporosis. Expert Opin. Pharmacother. 2020, 21, 721–732. [Google Scholar] [CrossRef]
- Lovsin, N.; Zupan, J.; Marc, J. Genetic effects on bone health. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kodric, K.; Zupan, J.; Kranjc, T.; Komadina, R.; Mlakar, V.; Marc, J.; Lovsin, N. Sex-determining region Y (SRY) attributes to gender differences in RANKL expression and incidence of osteoporosis. Exp. Mol. Med. 2019, 51, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hikita, A.; Yana, I.; Wakeyama, H.; Nakamura, M.; Kadono, Y.; Oshima, Y.; Nakamura, K.; Seiki, M.; Tanaka, S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J. Biol. Chem. 2006, 281, 36846–36855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Kasai, M.; Utsuyama, M.; Hirokawa, K. Determination of three isoforms of the receptor activator of nuclear factor-kappaB ligand and their differential expression in bone and thymus. Endocrinology 2001, 142, 1419–1426. [Google Scholar] [CrossRef]
- Nagai, M.; Kyakumoto, S.; Sato, N. Cancer cells responsible for humoral hypercalcemia express mRNA encoding a secreted form of ODF/TRANCE that induces osteoclast formation. Biochem. Biophys. Res. Commun. 2000, 269, 532–536. [Google Scholar] [CrossRef]
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Bartlett, F.S., 3rd; Frankel, W.N.; et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997, 272, 25190–25194. [Google Scholar] [CrossRef] [Green Version]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Fata, J.E.; Kong, Y.Y.; Li, J.; Sasaki, T.; Irie-Sasaki, J.; Moorehead, R.A.; Elliott, R.; Scully, S.; Voura, E.B.; Lacey, D.L.; et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 2000, 103, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- O’Brien, C.A. Control of RANKL Gene Expression. Bone 2010, 46, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dai, J.; Qi, Y.; Lin, D.L.; Smith, P.; Strayhorn, C.; Mizokami, A.; Fu, Z.; Westman, J.; Keller, E.T. Osteoprotegerin inhibits prostate cancer–induced osteoclastogenesis and prevents prostate tumor growth in the bone. J. Clin. Investig. 2001, 107, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Corey, E.; Lee, Z.D.; True, L.D.; Yun, T.J.; Tondravi, M.; Vessella, R.L. Osteoprotegerin and rank ligand expression in prostate cancer. Urology 2001, 57, 611–616. [Google Scholar] [CrossRef]
- Huang, L.; Cheng, Y.Y.; Chow, L.T.; Zheng, M.H.; Kumta, S.M. Tumour cells produce receptor activator of NF-kappaB ligand (RANKL) in skeletal metastases. J. Clin. Pathol. 2002, 55, 877–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, A.N.; Atkins, G.J.; To, L.B.; Pan, B.; Horvath, N.; Kostakis, P.; Findlay, D.M.; Bardy, P.; Zannettino, A.C. Receptor activator of nuclear factor-kappaB ligand expression by human myeloma cells mediates osteoclast formation in vitro and correlates with bone destruction in vivo. Cancer Res. 2003, 63, 5438–5445. [Google Scholar] [PubMed]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Gubrij, I.; Lin, S.C.; Saylors, R.L.; Manolagas, S.C. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-kappaB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J. Biol. Chem. 1999, 274, 19301–19308. [Google Scholar]
- Jilka, R.L.; Hangoc, G.; Girasole, G.; Passeri, G.; Williams, D.C.; Abrams, J.S.; Boyce, B.; Broxmeyer, H.; Manolagas, S.C. Increased osteoclast development after estrogen loss: Mediation by interleukin-6. Science 1992, 257, 88–91. [Google Scholar] [CrossRef]
- Holmen, S.L.; Zylstra, C.R.; Mukherjee, A.; Sigler, R.E.; Faugere, M.C.; Bouxsein, M.L.; Deng, L.; Clemens, T.L.; Williams, B.O. Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem. 2005, 280, 21162–21168. [Google Scholar] [CrossRef] [Green Version]
- Spencer, G.J.; Utting, J.C.; Etheridge, S.L.; Arnett, T.R.; Genever, P.G. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J. Cell Sci. 2006, 119, 1283–1296. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Janz, S. Attenuation of WNT signaling by DKK-1 and -2 regulates BMP2-induced osteoblast differentiation and expression of OPG, RANKL and M-CSF. Mol. Cancer 2007, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.S.; Ko, J.Y.; Lin, C.L.; Wu, H.L.; Ke, H.J.; Tai, P.J. Knocking down dickkopf-1 alleviates estrogen deficiency induction of bone loss. A histomorphological study in ovariectomized rats. Bone 2007, 40, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Kollet, O.; Heeschen, C.; Liebner, S.; Urbich, C.; Ihling, C.; Orlandi, A.; Lapidot, T.; Zeiher, A.M.; Dimmeler, S. The Wnt antagonist Dickkopf-1 mobilizes vasculogenic progenitor cells via activation of the bone marrow endosteal stem cell niche. Circ. Res. 2008, 103, 796–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Michigami, T.; Sakaguchi, N.; Kokubu, C.; Suzuki, A.; Namba, N.; Sakai, N.; Nakajima, S.; Imai, K.; Ozono, K. Lrp6 hypomorphic mutation affects bone mass through bone resorption in mice and impairs interaction with Mesd. J. Bone Miner. Res. 2008, 23, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.W.; Chen, Y.; Stephens, O.; Brown, N.; Chen, B.; Epstein, J.; Barlogie, B.; Shaughnessy, J.D., Jr. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: A potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 2008, 112, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Ozono, K.; Kubota, T.; Kondou, H.; Tachikawa, K.; Michigami, T. PTH/cAMP/PKA signaling facilitates canonical Wnt signaling via inactivation of glycogen synthase kinase-3beta in osteoblastic Saos-2 cells. J. Cell. Biochem. 2008, 104, 304–317. [Google Scholar] [CrossRef]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- Kassel, O.; Herrlich, P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol. Cell. Endocrinol. 2007, 275, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef]
- Hachemi, Y.; Rapp, A.E.; Picke, A.K.; Weidinger, G.; Ignatius, A.; Tuckermann, J. Molecular mechanisms of glucocorticoids on skeleton and bone regeneration after fracture. J. Mol. Endocrinol. 2018, 61, R75–R90. [Google Scholar] [CrossRef]
- Scheschowitsch, K.; Leite, J.A.; Assreuy, J. New Insights in Glucocorticoid Receptor Signaling-More Than Just a Ligand-Binding Receptor. Front. Endocrinol. (Lausanne) 2017, 8, 16. [Google Scholar] [CrossRef]
- Kondo, T.; Kitazawa, R.; Yamaguchi, A.; Kitazawa, S. Dexamethasone promotes osteoclastogenesis by inhibiting osteoprotegerin through multiple levels. J. Cell. Biochem. 2008, 103, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Koenen, M.; Schauer, S.; Wittig-Blaich, S.; Ahmad, M.; Baschant, U.; Tuckermann, J.P. Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol. Rev. 2016, 96, 409–447. [Google Scholar] [CrossRef] [Green Version]
- Harding, G.; Mak, Y.T.; Evans, B.; Cheung, J.; MacDonald, D.; Hampson, G. The effects of dexamethasone and dehydroepiandrosterone (DHEA) on cytokines and receptor expression in a human osteoblastic cell line: Potential steroid-sparing role for DHEA. Cytokine 2006, 36, 57–68. [Google Scholar] [CrossRef]
- Sivagurunathan, S.; Muir, M.M.; Brennan, T.C.; Seale, J.P.; Mason, R.S. Influence of glucocorticoids on human osteoclast generation and activity. J. Bone Miner. Res. 2005, 20, 390–398. [Google Scholar] [CrossRef]
- Rauner, M.; Goettsch, C.; Stein, N.; Thiele, S.; Bornhaeuser, M.; De Bosscher, K.; Haegeman, G.; Tuckermann, J.; Hofbauer, L.C. Dissociation of osteogenic and immunological effects by the selective glucocorticoid receptor agonist, compound A, in human bone marrow stromal cells. Endocrinology 2011, 152, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Zeitz, U.; Schoppet, M.; Skalicky, M.; Schuler, C.; Stolina, M.; Kostenuik, P.J.; Erben, R.G. Prevention of glucocorticoid-induced bone loss in mice by inhibition of RANKL. Arthritis Rheum. 2009, 60, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, R.; Kitazawa, S. Vitamin D(3) augments osteoclastogenesis via vitamin D-responsive element of mouse RANKL gene promoter. Biochem. Biophys. Res. Commun. 2002, 290, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Conaway, H.H.; Henning, P.; Lie, A.; Tuckermann, J.; Lerner, U.H. Glucocorticoids employ the monomeric glucocorticoid receptor to potentiate vitamin D3 and parathyroid hormone-induced osteoclastogenesis. FASEB J. 2019, 33, 14394–14409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Castro, J.; Garcia, R.; Garrido, P.; Isla, D.; Massuti, B.; Blanca, B.; Vazquez, J. Therapeutic Potential of Denosumab in Patients with Lung Cancer: Beyond Prevention of Skeletal Complications. Clin. Lung Cancer 2015, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 19, 2163–2171. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lovšin, N.; Marc, J. Glucocorticoid Receptor Regulates TNFSF11 Transcription by Binding to Glucocorticoid Responsive Element in TNFSF11 Proximal Promoter Region. Int. J. Mol. Sci. 2021, 22, 1054. https://doi.org/10.3390/ijms22031054
Lovšin N, Marc J. Glucocorticoid Receptor Regulates TNFSF11 Transcription by Binding to Glucocorticoid Responsive Element in TNFSF11 Proximal Promoter Region. International Journal of Molecular Sciences. 2021; 22(3):1054. https://doi.org/10.3390/ijms22031054
Chicago/Turabian StyleLovšin, Nika, and Janja Marc. 2021. "Glucocorticoid Receptor Regulates TNFSF11 Transcription by Binding to Glucocorticoid Responsive Element in TNFSF11 Proximal Promoter Region" International Journal of Molecular Sciences 22, no. 3: 1054. https://doi.org/10.3390/ijms22031054
APA StyleLovšin, N., & Marc, J. (2021). Glucocorticoid Receptor Regulates TNFSF11 Transcription by Binding to Glucocorticoid Responsive Element in TNFSF11 Proximal Promoter Region. International Journal of Molecular Sciences, 22(3), 1054. https://doi.org/10.3390/ijms22031054