
Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins in Yeast

Abstract
:1. Introduction
2. ER-Associated Degradation in Yeast
2.1. The Hrd1 Pathway
2.2. The Doa10 Pathway
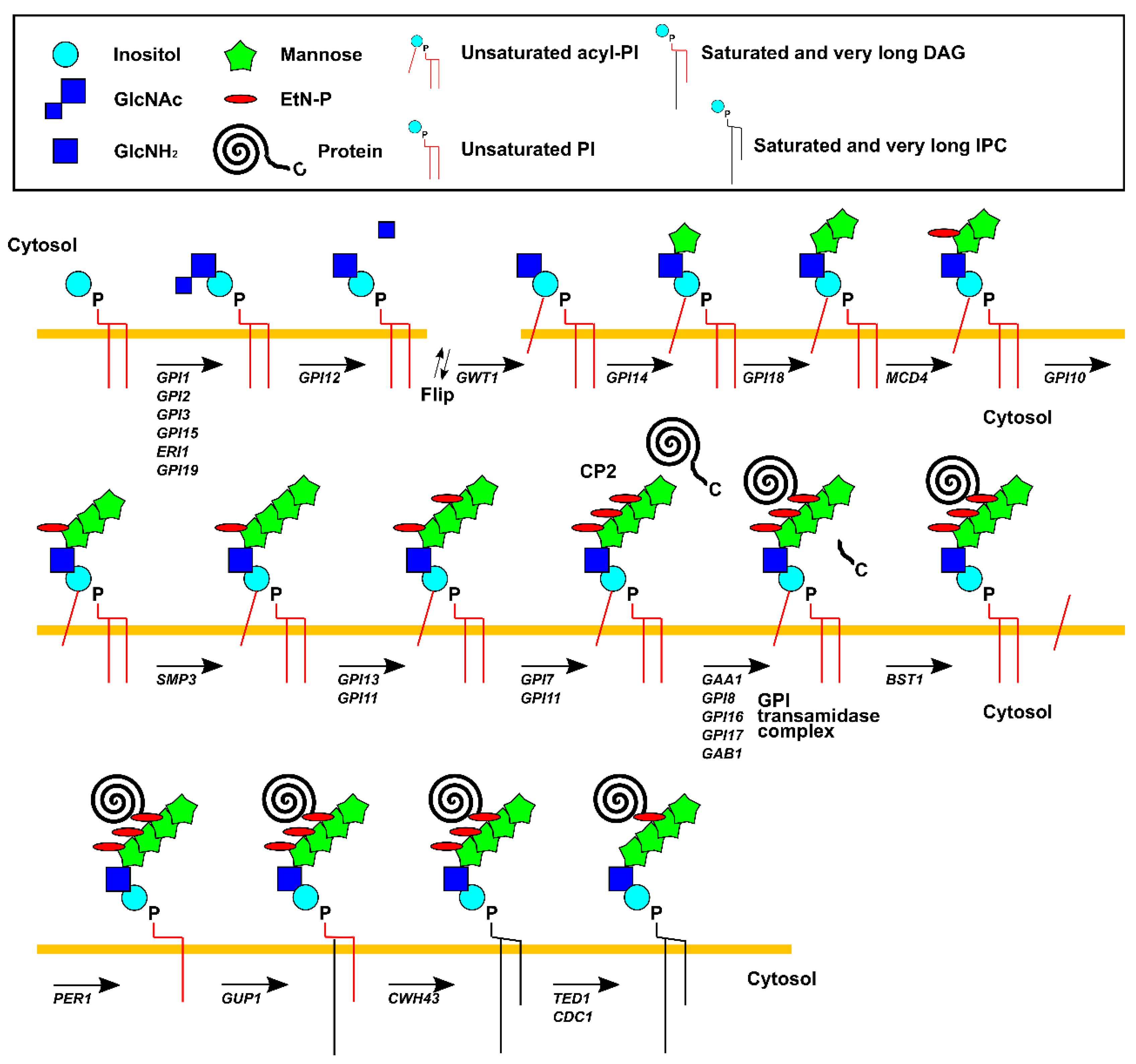
3. GPI-Anchored Proteins
4. Quality Control of GPI-Anchored Proteins
5. Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins
5.1. Genetic Interactions between GPI and ERAD Genes
5.2. Quality Control of Proteins that Harbor the GPI Anchoring Signal in the Cytosol
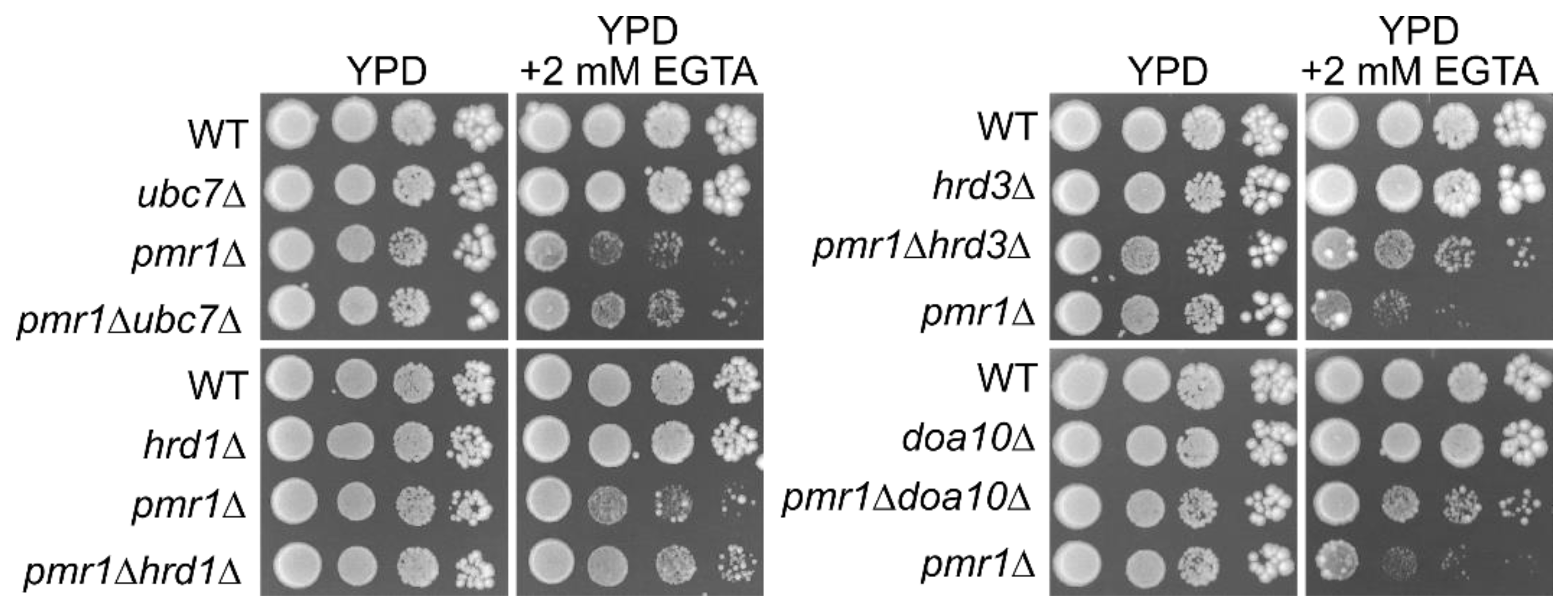
5.3. Exit of GPI-Anchored Proteins from the ER Is Affected by the Perturbation of Manganese Homeostasis
5.4. Possible Involvement of ERAD in the Maintenance of Manganese Homeostasis
6. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ERAD | Endoplasmic reticulum-associated degradation |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetra-acetic acid |
| GPI | Glycosylphosphatidylinositol |
| EtN-P | Ethanolamine phosphate |
| Man | Mannose |
| GlcN | Glucosamine |
| GlcNAc | N-Acetylglucosamine |
| UPR | Unfolded protein response |
| prERAD | Pre-insertional ERAD |
References
- Rüdiger, S.; Buchberger, A.; Bukau, B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Mol. Biol. 1997, 4, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [Green Version]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, C.; Hendershot, L.M. Disposing of misfolded ER proteins: A troubled substrate’s way out of the ER. Mol. Cell Endocrinol. 2020, 500, 110630. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Mehrtash, A.B.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin. Cell Dev. Biol. 2019, 93, 111–124. [Google Scholar] [CrossRef]
- Berner, N.; Reutter, K.-R.; Wolf, D.H. Protein Quality Control of the Endoplasmic Reticulum and Ubiquitin–Proteasome-Triggered Degradation of Aberrant Proteins: Yeast Pioneers the Path. Annu. Rev. Biochem. 2018, 87, 751–782. [Google Scholar] [CrossRef]
- Christianson, J.C.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Nakatsukasa, K.; Brodsky, J.L. The Recognition and Retrotranslocation of Misfolded Proteins from the Endoplasmic Reticulum. Traffic 2008, 9, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Nakatsukasa, K.; Okumura, F.; Kamura, T. Proteolytic regulation of metabolic enzymes by E3 ubiquitin ligase complexes: Lessons from yeast. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Tsai, B.; Arvan, P. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef]
- Adle, D.J.; Wei, W.; Smith, N.; Bies, J.J.; Lee, J. Cadmium-mediated rescue from ER-associated degradation induces expression of its exporter. Proc. Natl. Acad. Sci. USA 2009, 106, 10189–10194. [Google Scholar] [CrossRef] [Green Version]
- Hampton, R.Y.; Gardner, R.G.; Rine, J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 1996, 7, 2029–2044. [Google Scholar] [CrossRef] [Green Version]
- Rape, M.; Hoppe, T.; Gorr, I.; Kalocay, M.; Richly, H.; Jentsch, S. Mobilization of Processed, Membrane-Tethered SPT23 Transcription Factor by CDC48UFD1/NPL4, a Ubiquitin-Selective Chaperone. Cell 2001, 107, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Foresti, O.; Ruggiano, A.; Hannibal-Bach, H.K.; Ejsing, C.S.; Carvalho, P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2013, 2, e00953. [Google Scholar] [CrossRef]
- Wu, X.; Siggel, M.; Ovchinnikov, S.; Mi, W.; Svetlov, V.; Nudler, E.; Liao, M.; Hummer, G.; Rapoport, T.A. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 2020, 368, eaaz2449. [Google Scholar] [CrossRef]
- Knop, M.; Finger, A.; Braun, T.; Hellmuth, K.; Wolf, D.H. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996, 15, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, J.; Plemper, R.K.; Finger, A.; Wolf, D.H. Der3p/Hrd1p Is Required for Endoplasmic Reticulum-associated Degradation of Misfolded Lumenal and Integral Membrane Proteins. Mol. Biol. Cell 1998, 9, 209–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, N.W.; Gardner, R.G.; Seelig, L.P.; Joazeiro, C.A.; Hampton, R.Y. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 2001, 3, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Gauss, R.; Jarosch, E.; Sommer, T.; Hirsch, C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat. Cell Biol. 2006, 8, 849–854. [Google Scholar] [CrossRef]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct Ubiquitin-Ligase Complexes Define Convergent Pathways for the Degradation of ER Proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Kanehara, K.; Xie, W.; Ng, D.T.W. Modularity of the Hrd1 ERAD complex underlies its diverse client range. J. Cell Biol. 2010, 188, 707–716. [Google Scholar] [CrossRef]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Gauss, R.; Sommer, T.; Jarosch, E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006, 25, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Bhamidipati, A.; Denic, V.; Quan, E.M.; Weissman, J.S. Exploration of the Topological Requirements of ERAD Identifies Yos9p as a Lectin Sensor of Misfolded Glycoproteins in the ER Lumen. Mol. Cell 2005, 19, 741–751. [Google Scholar] [CrossRef]
- Szathmary, R.; Bielmann, R.; Nita-Lazar, M.; Burda, P.; Jakob, C.A. Yos9 Protein Is Essential for Degradation of Misfolded Glycoproteins and May Function as Lectin in ERAD. Mol. Cell 2005, 19, 765–775. [Google Scholar] [CrossRef]
- Quan, E.M.; Kamiya, Y.; Kamiya, D.; Denic, V.; Weibezahn, J.; Kato, K.; Weissman, J.S. Defining the Glycan Destruction Signal for Endoplasmic Reticulum-Associated Degradation. Mol. Cell 2008, 32, 870–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, C.A.; Bodmer, D.; Spirig, U.; Bättig, P.; Marcil, A.; Dignard, D.; Bergeron, J.J.; Thomas, D.Y.; Aebi, M. Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2001, 2, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsukasa, K.; Nishikawa, S.-I.; Hosokawa, N.; Nagata, K.; Endo, T. Mnl1p, an α-Mannosidase-like Protein in YeastSaccharomyces cerevisiae, Is Required for Endoplasmic Reticulum-associated Degradation of Glycoproteins. J. Biol. Chem. 2001, 276, 8635–8638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Kanehara, K.; Sayeed, A.; Ng, D.T.W. Intrinsic Conformational Determinants Signal Protein Misfolding to the Hrd1/Htm1 Endoplasmic Reticulum–associated Degradation System. Mol. Biol. Cell 2009, 20, 3317–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashistha, N.; Neal, S.E.; Singh, A.; Carroll, S.M.; Hampton, R.Y. Direct and essential function for Hrd3 in ER-associated degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 5934–5939. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.C.; Hanna, J.; Hirsch, C.; Volkwein, C.; Schütz, A.; Heinemann, U.; Sommer, T.; Jarosch, E. Usa1 Functions as a Scaffold of the HRD-Ubiquitin Ligase. Mol. Cell 2009, 36, 782–793. [Google Scholar] [CrossRef]
- Biederer, T.; Volkwein, C.; Sommer, T. Role of Cue1p in Ubiquitination and Degradation at the ER Surface. Science 1997, 278, 1806–1809. [Google Scholar] [CrossRef] [Green Version]
- Schuberth, C.; Buchberger, A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 999–1006. [Google Scholar] [CrossRef]
- Neuber, O.; Jarosch, E.; Volkwein, C.; Walter, J.; Sommer, T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 993–998. [Google Scholar] [CrossRef]
- Nakatsukasa, K.; Brodsky, J.L.; Kamura, T. A stalled retrotranslocation complex reveals physical linkage between substrate recognition and proteasomal degradation during ER-associated degradation. Mol. Biol. Cell 2013, 24, 1765–1775. [Google Scholar] [CrossRef]
- Sato, B.K.; Schulz, D.; Do, P.H.; Hampton, R.Y. Misfolded Membrane Proteins Are Specifically Recognized by the Transmembrane Domain of the Hrd1p Ubiquitin Ligase. Mol. Cell 2009, 34, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, S.; Jaeger, P.A.; Duttke, S.H.; Benner, C.; Glass, C.K.; Ideker, T.; Hampton, R.Y. The Dfm1 Derlin Is Required for ERAD Retrotranslocation of Integral Membrane Proteins. Mol. Cell 2018, 69, 306–320.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, S.E.; Syau, D.; Nejatfard, A.; Nadeau, S.; Hampton, R.Y. HRD Complex Self-Remodeling Enables a Novel Route of Membrane Protein Retrotranslocation. iScience 2020, 23, 101493. [Google Scholar] [CrossRef] [PubMed]
- Kreft, S.G.; Wang, L.; Hochstrasser, M. Membrane Topology of the Yeast Endoplasmic Reticulum-localized Ubiquitin Ligase Doa10 and Comparison with Its Human Ortholog TEB4 (MARCH-VI). J. Biol. Chem. 2006, 281, 4646–4653. [Google Scholar] [CrossRef] [Green Version]
- Huyer, G.; Piluek, W.F.; Fansler, Z.; Kreft, S.G.; Hochstrasser, M.; Brodsky, J.L.; Michaelis, S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 2004, 279, 38369–38378. [Google Scholar] [CrossRef] [Green Version]
- Gnann, A.; Riordan, J.R.; Wolf, D.H. Cystic Fibrosis Transmembrane Conductance Regulator Degradation Depends on the Lectins Htm1p/EDEM and the Cdc48 Protein Complex in Yeast. Mol. Biol. Cell 2004, 15, 4125–4135. [Google Scholar] [CrossRef] [Green Version]
- Ravid, T.; Kreft, S.G.; Hochstrasser, M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006, 25, 533–543. [Google Scholar] [CrossRef]
- Vashist, S.; Ng, D.T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004, 165, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Furth, N.; Gertman, O.; Shiber, A.; Alfassy, O.S.; Cohen, I.; Rosenberg, M.M.; Doron, N.K.; Friedler, A.; Ravid, T. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol. Biol. Cell 2011, 22, 4726–4739. [Google Scholar] [CrossRef]
- Swanson, R.; Locher, M.; Hochstrasser, M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001, 15, 2660–2674. [Google Scholar] [CrossRef] [Green Version]
- Habeck, G.; Ebner, F.A.; Shimada-Kreft, H.; Kreft, S.G. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J. Cell Biol. 2015, 209, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.C.; Vasic, V.; Stein, A. Doa10 is a membrane protein retrotranslocase in ER-associated protein degradation. eLife 2020, 9, e56945. [Google Scholar] [CrossRef] [PubMed]
- Nakatsukasa, K.; Huyer, G.; Michaelis, S.; Brodsky, J.L. Dissecting the ER-Associated Degradation of a Misfolded Polytopic Membrane Protein. Cell 2008, 132, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Liu, Y.; Chang, A. Cytoplasmic Hsp70 Promotes Ubiquitination for Endoplasmic Reticulum-associated Degradation of a Misfolded Mutant of the Yeast Plasma Membrane ATPase, PMA1. J. Biol. Chem. 2007, 282, 26140–26149. [Google Scholar] [CrossRef] [Green Version]
- Metzger, M.B.; Maurer, M.J.; Dancy, B.M.; Michaelis, S. Degradation of a Cytosolic Protein Requires Endoplasmic Reticulum-associated Degradation Machinery. J. Biol. Chem. 2008, 283, 32302–32316. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Cohen, I.; Popp, O.; Dittmar, G.; Reiss, Y.; Sommer, T.; Ravid, T.; Jarosch, E. Sequential Poly-ubiquitylation by Specialized Conjugating Enzymes Expands the Versatility of a Quality Control Ubiquitin Ligase. Mol. Cell 2016, 63, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Nakatsukasa, K.; Kamura, T. Subcellular Fractionation Analysis of the Extraction of Ubiquitinated Polytopic Membrane Substrate during ER-Associated Degradation. PLoS ONE 2016, 11, e0148327. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Urban, J.; Volkwein, C.; Sommer, T. Sec61p-independent degradation of the tail-anchored ER membrane protein Ubc6p. EMBO J. 2001, 20, 3124–3131. [Google Scholar] [CrossRef] [Green Version]
- Pittet, M.; Conzelmann, A. Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Leidich, S.D.; Kostova, Z.; Latek, R.R.; Costello, L.C.; Drapp, D.A.; Gray, W.; Fassler, J.S.; Orlean, P. Temperature-sensitive Yeast GPI Anchoring Mutants gpi2 and gpi3 Are Defective in the Synthesis of N-Acetylglucosaminyl Phosphatidylinositol. J. Biol. Chem. 1995, 270, 13029–13035. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.C.; Westfall, B.A.; Orlean, P. Ynl038wp (Gpi15p) is theSaccharomyces cerevisiae homologue of human Pig-Hp and participates in the first step in glycosylphosphatidylinositol assembly. Yeast 2001, 18, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, H.A.; Romeo, M.J.; Lewis, S.E.; Yan, B.C.; Orlean, P.; Levin, D.E. Gpi19, the Saccharomyces cerevisiae Homologue of Mammalian PIG-P, Is a Subunit of the Initial Enzyme for Glycosylphosphatidylinositol Anchor Biosynthesis. Eukaryot. Cell 2005, 4, 1801–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlean, P.; Menon, A.K. Thematic review series: Lipid Posttranslational Modifications.GPI anchoring of protein in yeast and mammalian cells, or: How we learned to stop worrying and love glycophospholipids. J. Lipid Res. 2007, 48, 993–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, Y.; Watanabe, R.; Harris, C.L.; Hong, Y.; Ohishi, K.; Kinoshita, K.; Kinoshita, T. PIG-M transfers the first mannose to glycosylphosphatidylinositol on the lumenal side of the ER. EMBO J. 2001, 20, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.Y.; Hong, Y.; Ashida, H.; Shishioh, N.; Murakami, Y.; Morita, Y.S.; Maeda, Y.; Kinoshita, T. PIG-V Involved in Transferring the Second Mannose in Glycosylphosphatidylinositol. J. Biol. Chem. 2005, 280, 9489–9497. [Google Scholar] [CrossRef] [Green Version]
- Gaynor, E.C.; Mondésert, G.; Grimme, S.J.; Reed, S.I.; Orlean, P.; Emr, S.D. MCD4Encodes a Conserved Endoplasmic Reticulum Membrane Protein Essential for Glycosylphosphatidylinositol Anchor Synthesis in Yeast. Mol. Biol. Cell 1999, 10, 627–648. [Google Scholar] [CrossRef] [Green Version]
- Sütterlin, C.; Escribano, M.V.; Gerold, P.; Maeda, Y.; Mazón, M.J.; Kinoshita, T.; Schwarz, R.T.; Riezman, H. Saccharomyces cerevisiae GPI10, the functional homologue of human PIG-B, is required for glycosylphosphatidylinositol-anchor synthesis. Biochem. J. 1998, 332, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Flury, I.; Benachour, A.; Conzelmann, A. YLL031c Belongs to a Novel Family of Membrane Proteins Involved in the Transfer of Ethanolaminephosphate onto the Core Structure of Glycosylphosphatidylinositol Anchors in Yeast. J. Biol. Chem. 2000, 275, 24458–24465. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.J.; Westfall, B.A.; Wiedman, J.M.; Taron, C.H.; Orlean, P. The Essential Smp3 Protein Is Required for Addition of the Side-branching Fourth Mannose during Assembly of Yeast Glycosylphosphatidylinositols. J. Biol. Chem. 2001, 276, 27731–27739. [Google Scholar] [CrossRef] [Green Version]
- Benachour, A.; Sipos, G.; Flury, I.; Reggiori, F.; Canivenc-Gansel, E.; Vionnet, C.; Conzelmann, A.; Benghezal, M. Deletion of GPI7, a Yeast Gene Required for Addition of a Side Chain to the Glycosylphosphatidylinositol (GPI) Core Structure, Affects GPI Protein Transport, Remodeling, and Cell Wall Integrity. J. Biol. Chem. 1999, 274, 15251–15261. [Google Scholar] [CrossRef] [Green Version]
- Imhof, I.; Flury, I.; Vionnet, C.; Roubaty, C.; Egger, D.; Conzelmann, A. Glycosylphosphatidylinositol (GPI) Proteins ofSaccharomyces cerevisiaeContain Ethanolamine Phosphate Groups on the α1,4-linked Mannose of the GPI Anchor. J. Biol. Chem. 2004, 279, 19614–19627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Knez, J.J.; Merrick, W.C.; Medof, M.E. Comparative efficiencies of C-terminal signals of native glycophosphatidylinositol (GPI)-anchored proproteins in conferring GPI-anchoring. J. Cell. Biochem. 2002, 84, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Inoue, N.; Kinoshita, T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 2001, 20, 4088–4098. [Google Scholar] [CrossRef]
- Benghezal, M.; Benachour, A.; Rusconi, S.; Aebi, M.; Conzelmann, A. Yeast Gpi8p is essential for GPI anchor attachment onto proteins. EMBO J. 1996, 15, 6575–6583. [Google Scholar] [CrossRef] [PubMed]
- Fraering, P.; Imhof, I.; Meyer, U.; Strub, J.-M.; Van Dorsselaer, A.; Vionnet, C.; Conzelmann, A. The GPI Transamidase Complex of Saccharomyces cerevisiae Contains Gaa1p, Gpi8p, and Gpi16p. Mol. Biol. Cell 2001, 12, 3295–3306. [Google Scholar] [CrossRef] [Green Version]
- Hamburger, D.; Egerton, M.; Riezman, H. Yeast Gaa1p is required for attachment of a completed GPI anchor onto proteins. J. Cell Biol. 1995, 129, 629–639. [Google Scholar] [CrossRef]
- Hong, Y.; Ohishi, K.; Kang, J.Y.; Tanaka, S.; Inoue, N.; Nishimura, J.-I.; Maeda, Y.; Kinoshita, T. Human PIG-U and Yeast Cdc91p Are the Fifth Subunit of GPI Transamidase That Attaches GPI-Anchors to Proteins. Mol. Biol. Cell 2003, 14, 1780–1789. [Google Scholar] [CrossRef] [Green Version]
- Meyer, U.; Benghezal, M.; Imhof, I.; Conzelmann, A. Active Site Determination of Gpi8p, a Caspase-Related Enzyme Required for Glycosylphosphatidylinositol Anchor Addition to Proteins†. Biochemistry 2000, 39, 3461–3471. [Google Scholar] [CrossRef]
- Ohishi, K.; Inoue, N.; Maeda, Y.; Takeda, J.; Riezman, H.; Kinoshita, T. Gaa1p and Gpi8p Are Components of a Glycosylphosphatidylinositol (GPI) Transamidase That Mediates Attachment of GPI to Proteins. Mol. Biol. Cell 2000, 11, 1523–1533. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Szallies, A.; Rawer, M.; Echner, H.; Duszenko, M. GPI anchor transamidase of Trypanosoma brucei: In vitro assay of the recombinant protein and VSG anchor exchange. J. Cell Sci. 2002, 115, 2529–2539. [Google Scholar]
- Tanaka, S.; Maeda, Y.; Tashima, Y.; Kinoshita, T. Inositol Deacylation of Glycosylphosphatidylinositol-anchored Proteins Is Mediated by Mammalian PGAP1 and Yeast Bst1p. J. Biol. Chem. 2004, 279, 14256–14263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Umemura, M.; Yoko-o, T.; Jigami, Y. PER1 Is Required for GPI-Phospholipase A2 Activity and Involved in Lipid Remodeling of GPI-anchored Proteins. Mol. Biol. Cell 2006, 17, 5253–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosson, R.; Jaquenoud, M.; Conzelmann, A. GUP1 of Saccharomyces cerevisiae Encodes an O-Acyltransferase Involved in Remodeling of the GPI Anchor. Mol. Biol. Cell 2006, 17, 2636–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, M.; Fujita, M.; Yoko-o, T.; Fukamizu, A.; Jigami, Y. Saccharomyces cerevisiae CWH43Is Involved in the Remodeling of the Lipid Moiety of GPI Anchors to Ceramides. Mol. Biol. Cell 2007, 18, 4304–4316. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, H.M.; Vionnet, C.; Roubaty, C.; Conzelmann, A. Cdc1 removes the ethanolamine phosphate of the first mannose of GPI anchors and thereby facilitates the integration of GPI proteins into the yeast cell wall. Mol. Biol. Cell 2014, 25, 3375–3388. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Lopez, J.; Perez-Linero, A.M.; Aguilera-Romero, A.; Martin, M.E.; Okano, T.; Silva, D.V.; Seeberger, P.H.; Riezman, H.; Funato, K.; Goder, V.; et al. COPII Coat Composition Is Actively Regulated by Luminal Cargo Maturation. Curr. Biol. 2015, 25, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Maeda, Y.; Ra, M.; Yamaguchi, Y.; Taguchi, R.; Kinoshita, T. GPI Glycan Remodeling by PGAP5 Regulates Transport of GPI-Anchored Proteins from the ER to the Golgi. Cell 2009, 139, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Yoko-o, T.; Jigami, Y. Inositol Deacylation by Bst1p Is Required for the Quality Control of Glycosylphosphatidylinositol-anchored Proteins. Mol. Biol. Cell 2006, 17, 834–850. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, H.; Fujita, M.; Yoko-o, T.; Jigami, Y. O-Mannosylation is Required for Degradation of the Endoplasmic Reticulum-associated Degradation Substrate Gas1*p via the Ubiquitin/Proteasome Pathway in Saccharomyces cerevisiae. J. Biochem. 2007, 143, 555–567. [Google Scholar] [CrossRef]
- Marzioch, M.; Henthorn, D.C.; Herrmann, J.M.; Wilson, R.; Thomas, D.Y.; Bergeron, J.J.; Solari, R.C.; Rowley, A. Erp1p and Erp2p, partners for Emp24p and Erv25p in a yeast p24 complex. Mol. Biol. Cell 1999, 10, 1923–1938. [Google Scholar] [CrossRef] [Green Version]
- Ashok, A.; Hegde, R.S. Selective Processing and Metabolism of Disease-Causing Mutant Prion Proteins. PLoS Pathog. 2009, 5, e1000479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satpute-Krishnan, P.; Ajinkya, M.; Bhat, S.; Itakura, E.; Hegde, R.S.; Lippincott-Schwartz, J. ER Stress-Induced Clearance of Misfolded GPI-Anchored Proteins via the Secretory Pathway. Cell 2014, 158, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorska, N.; Lemus, L.; Romero, M.A.A.; Manzano-Lopez, J.; Riezman, H.; Muñiz, M.; Goder, V. Limited ER quality control for GPI-anchored proteins. J. Cell Biol. 2016, 213, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Banfield, D.K. Cdc1p is a Golgi-localized Glycosylphosphatidylinositol-anchored Protein Remodelase. Mol. Biol. Cell 2020, 31, 2883–2891. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Hegde, R.S. Misfolded GPI-anchored proteins are escorted through the secretory pathway by ER-derived factors. eLife 2019, 8, e46740. [Google Scholar] [CrossRef]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, aaf1420. [Google Scholar] [CrossRef]
- Costanzo, M.; Kuzmin, E.; Van Leeuwen, J.; Mair, B.; Moffat, J.; Boone, C.; Andrews, B.J. Global Genetic Networks and the Genotype-to-Phenotype Relationship. Cell 2019, 177, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Maeda, Y.; Liu, Y.-S.; Takada, Y.; Ninomiya, A.; Hirata, T.; Fujita, M.; Murakami, Y.; Kinoshita, T. Cross-talks of glycosylphosphatidylinositol biosynthesis with glycosphingolipid biosynthesis and ER-associated degradation. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ast, T.; Cohen, G.; Schuldiner, M. A Network of Cytosolic Factors Targets SRP-Independent Proteins to the Endoplasmic Reticulum. Cell 2013, 152, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Hegde, R.S. A Calmodulin-Dependent Translocation Pathway for Small Secretory Proteins. Cell 2011, 147, 1576–1588. [Google Scholar] [CrossRef] [Green Version]
- Levine, C.G.; Mitra, D.; Sharma, A.; Smith, C.L.; Hegde, R.S. The Efficiency of Protein Compartmentalization into the Secretory Pathway. Mol. Biol. Cell 2005, 16, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Ast, T.; Aviram, N.; Chuartzman, S.G.; Schuldiner, M. A cytosolic degradation pathway, prERAD, monitors pre-inserted secretory pathway proteins. J. Cell Sci. 2014, 127, 3017–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paidhungat, M.; Garrett, S. Cdc1 and the vacuole coordinately regulate Mn2+ homeostasis in the yeast Saccharomyces cerevisiae. Genetics 1998, 148, 1787–1798. [Google Scholar] [PubMed]
- Čopič, A.; Dorrington, M.; Pagant, S.; Barry, J.; Lee, M.C.S.; Singh, I.; Hartman, J.L.; Miller, E.A. Genomewide Analysis Reveals Novel Pathways Affecting Endoplasmic Reticulum Homeostasis, Protein Modification and Quality Control. Genetics 2009, 182, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Haass, F.A.; Jonikas, M.; Walter, P.; Weissman, J.S.; Jan, Y.-N.; Jan, L.Y.; Schuldiner, M. Identification of yeast proteins necessary for cell-surface function of a potassium channel. Proc. Natl. Acad. Sci. USA 2007, 104, 18079–18084. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.; Megyeri, M.; Chen, O.C.W.; Condomitti, G.; Riezman, I.; Loizides-Mangold, U.; Abdul-Sada, A.; Rimon, N.; Riezman, H.; Platt, F.M.; et al. The Yeast P5 Type ATPase, Spf1, Regulates Manganese Transport into the Endoplasmic Reticulum. PLoS ONE 2013, 8, e85519. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, H.K.; Antebi, A.; Fink, G.R.; Buckley, C.M.; Dorman, T.E.; Levitre, J.; Davidow, L.S.; Mao, J.-I.; Moir, D.T. The yeast secretory pathway is perturbed by mutations in PMR1, a member of a Ca2+ ATPase family. Cell 1989, 58, 133–145. [Google Scholar] [CrossRef]
- Ton, V.-K.; Mandal, D.; Vahadji, C.; Rao, R. Functional Expression in Yeast of the Human Secretory Pathway Ca2+, Mn2+-ATPase Defective in Hailey-Hailey Disease. J. Biol. Chem. 2002, 277, 6422–6427. [Google Scholar] [CrossRef] [Green Version]
- Missiaen, L.; Raeymaekers, L.; Dode, L.; Vanoevelen, J.; Van Baelen, K.; Parys, J.B.; Callewaert, G.; De Smedt, H.; Segaert, S.; Wuytack, F. SPCA1 pumps and Hailey–Hailey disease. Biochem. Biophys. Res. Commun. 2004, 322, 1204–1213. [Google Scholar] [CrossRef]
- Xiang, M.; Mohamalawari, D.; Rao, R. A Novel Isoform of the Secretory Pathway Ca2+,Mn2+-ATPase, hSPCA2, Has Unusual Properties and Is Expressed in the Brain. J. Biol. Chem. 2005, 280, 11608–11614. [Google Scholar] [CrossRef] [Green Version]
- Antebi, A.; Fink, G.R. The yeast Ca(2+)-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol. Biol. Cell 1992, 3, 633–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dürr, G.; Strayle, J.; Plemper, R.; Elbs, S.; Klee, S.K.; Catty, P.; Wolf, D.H.; Rudolph, H.K. The medial-Golgi Ion Pump Pmr1 Supplies the Yeast Secretory Pathway with Ca2+ and Mn2+ Required for Glycosylation, Sorting, and Endoplasmic Reticulum-Associated Protein Degradation. Mol. Biol. Cell 1998, 9, 1149–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorin, A.; Rosas, G.; Rao, R. PMR1, a Ca2+-ATPase in Yeast Golgi, Has Properties Distinct from Sarco/endoplasmic Reticulum and Plasma Membrane Calcium Pumps. J. Biol. Chem. 1997, 272, 9895–9901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Culotta, V.C.; Yang, M.; Hall, M.D. Manganese Transport and Trafficking: Lessons Learned from Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Vashist, S.; Frank, C.G.; Jakob, C.A.; Ng, D.T. Two Distinctly Localized P-Type ATPases Collaborate to Maintain Organelle Homeostasis Required for Glycoprotein Processing and Quality Control. Mol. Biol. Cell 2002, 13, 3955–3966. [Google Scholar] [CrossRef] [Green Version]
- Kellermayer, R. Hailey-Hailey disease as an orthodisease of PMR1 deficiency inSaccharomyces cerevisiae. FEBS Lett. 2005, 579, 2021–2025. [Google Scholar] [CrossRef] [Green Version]
- Jonikas, M.C.; Collins, S.R.; Denic, V.; Oh, E.; Quan, E.M.; Schmid, V.; Weibezahn, J.; Schwappach, B.; Walter, P.; Weissman, J.S.; et al. Comprehensive Characterization of Genes Required for Protein Folding in the Endoplasmic Reticulum. Science 2009, 323, 1693–1697. [Google Scholar] [CrossRef] [Green Version]
- Paidhungat, M.; Garrett, S. Cdc1 is required for growth and Mn2+ regulation in Saccharomyces cerevisiae. Genetics 1998, 148, 1777–1786. [Google Scholar]
- Losev, E.; Papanikou, E.; Rossanese, O.W.; Glick, B.S. Cdc1p Is an Endoplasmic Reticulum-Localized Putative Lipid Phosphatase That Affects Golgi Inheritance and Actin Polarization by Activating Ca2+ Signaling. Mol. Cell. Biol. 2008, 28, 3336–3343. [Google Scholar] [CrossRef] [Green Version]
- Sahu, P.K.; Tomar, R.S. The natural anticancer agent cantharidin alters GPI-anchored protein sorting by targeting Cdc1-mediated remodeling in endoplasmic reticulum. J. Biol. Chem. 2019, 294, 3837–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| GPI2 | GPI8 | GPI10 | GPI11 | GPI13 | GPI16 | GPI17 | GPI19 | |
|---|---|---|---|---|---|---|---|---|
| HRD1 | P | P | P | P | ||||
| HRD3 | P | P | P | N | ||||
| UBC7 | P | P | P | |||||
| USA1 | P | P | P | P | ||||
| DER1 | N | P | P | |||||
| YOS9 | P | N | N | |||||
| DOA10 | P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakatsukasa, K. Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins in Yeast. Int. J. Mol. Sci. 2021, 22, 1061. https://doi.org/10.3390/ijms22031061
Nakatsukasa K. Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins in Yeast. International Journal of Molecular Sciences. 2021; 22(3):1061. https://doi.org/10.3390/ijms22031061
Chicago/Turabian StyleNakatsukasa, Kunio. 2021. "Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins in Yeast" International Journal of Molecular Sciences 22, no. 3: 1061. https://doi.org/10.3390/ijms22031061
APA StyleNakatsukasa, K. (2021). Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins in Yeast. International Journal of Molecular Sciences, 22(3), 1061. https://doi.org/10.3390/ijms22031061