
Changing Functional Signatures of Microglia along the Axis of Brain Aging



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Functional Signature of Young Adult Microglia
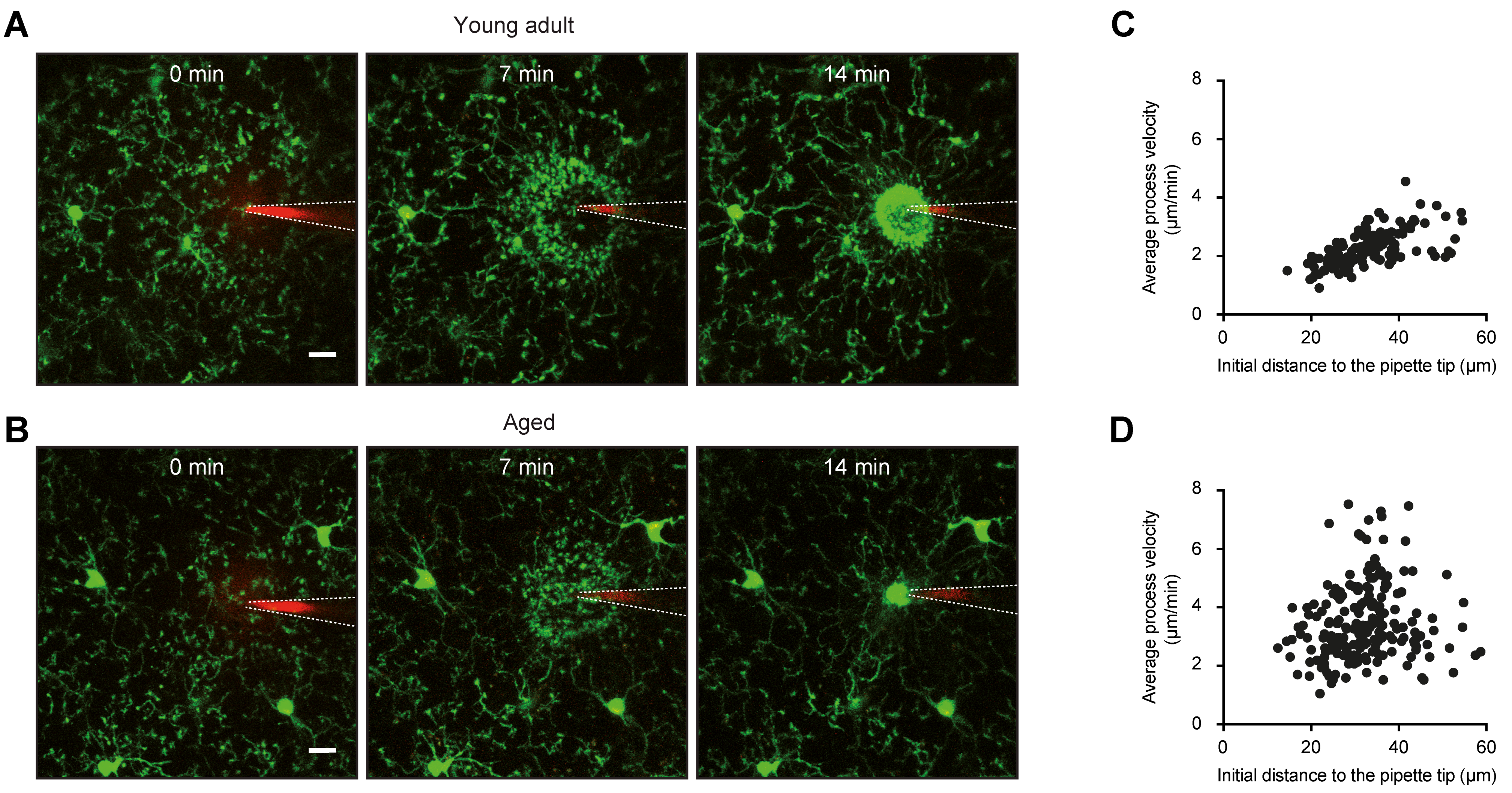
2.1. Morphology and Surveillance
2.2. Ca2+ Signaling
2.3. Gender-Specific Differences
3. Unexpected Alertness of Middle-Age Microglia
4. Functional Signatures of Aged Microglia
4.1. Transcriptional Signatures of Aged Microglia
4.2. Morphology and Surveillance
4.3. Ca2+ Signaling
4.4. Gender-Specific Differences
5. Counteracting Aging of Microglia
5.1. Enhancing the α7 nAChR Function
5.2. Effect of Caloric Restriction
5.3. The Role of Physical Exercise
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drachman, D.A. Aging of the brain, entropy, and Alzheimer disease. Neurology 2006, 67, 1340–1352. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Tichauer, J.E.; Eugenin, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 2010, 112, 1099–1114. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and Garb-aging. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]
- Garaschuk, O.; Semchyshyn, H.M.; Lushchak, V.I. Healthy brain aging: Interplay between reactive species, inflammation and energy supply. Ageing Res. Rev. 2018, 43, 26–45. [Google Scholar] [CrossRef]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [Green Version]
- von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Brawek, B.; Garaschuk, O. Microglial calcium signaling in the adult, aged and diseased brain. Cell Calcium 2013, 53, 159–169. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar] [CrossRef]
- Garaschuk, O. Age-related changes in microglial physiology: The role for healthy brain ageing and neurodegenerative disorders. Neuroforum 2017, 23, A182–A191. [Google Scholar]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Sankowski, R.; Bottcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Villa, A.; Della Torre, S.; Maggi, A. Sexual differentiation of microglia. Front. Neuroendocrinol. 2019, 52, 156–164. [Google Scholar] [CrossRef]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Flurkey, K.; Currer, J.M.; Harrison, D.E. The mouse in aging research. In The Mouse in Biomedical Research, 2nd ed.; Fox, J.G., Barthold, S., Davisson, M., Newcomer, C., Quimby, F., Smith, A., Eds.; American College Laboratory Animal Medicine (Elsevier): Burlington, MA, USA, 2007; pp. 6637–6672. [Google Scholar]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Eyo, U.B.; Peng, J.; Swiatkowski, P.; Mukherjee, A.; Bispo, A.; Wu, L.J. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. 2014, 34, 10528–10540. [Google Scholar] [CrossRef] [Green Version]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Tvrdik, P.; Kalani, M.Y.S. In Vivo Imaging of Microglial Calcium Signaling in Brain Inflammation and Injury. Int. J. Mol. Sci. 2017, 18, 2366. [Google Scholar] [CrossRef] [Green Version]
- Brawek, B.; Garaschuk, O. Monitoring in vivo function of cortical microglia. Cell Calcium 2017, 64, 109–117. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Madry, C.; Kyrargyri, V.; Arancibia-Carcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1beta Release Are Regulated by the Two-Pore Domain K(+) Channel THIK-1. Neuron 2018, 97, 299–312. [Google Scholar] [CrossRef]
- Dissing-Olesen, L.; LeDue, J.M.; Rungta, R.L.; Hefendehl, J.K.; Choi, H.B.; MacVicar, B.A. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 2014, 34, 10511–10527. [Google Scholar] [CrossRef] [Green Version]
- Umpierre, A.D.; Bystrom, L.L.; Ying, Y.; Liu, Y.U.; Worrell, G.; Wu, L.J. Microglial calcium signaling is attuned to neuronal activity in awake mice. eLife 2020, 9. [Google Scholar] [CrossRef]
- Liu, Y.U.; Ying, Y.; Li, Y.; Eyo, U.B.; Chen, T.; Zheng, J.; Umpierre, A.D.; Zhu, J.; Bosco, D.B.; Dong, H.; et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci. 2019, 22, 1771–1781. [Google Scholar] [CrossRef]
- Stowell, R.D.; Sipe, G.O.; Dawes, R.P.; Batchelor, H.N.; Lordy, K.A.; Whitelaw, B.S.; Stoessel, M.B.; Bidlack, J.M.; Brown, E.; Sur, M.; et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci. 2019, 22, 1782–1792. [Google Scholar] [CrossRef]
- Eichhoff, G.; Brawek, B.; Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim. Biophys. Acta 2011, 1813, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Schwendele, B.; Brawek, B.; Hermes, M.; Garaschuk, O. High-resolution in vivo imaging of microglia using a versatile nongenetically encoded marker. Eur. J. Immunol. 2012, 42, 2193–2196. [Google Scholar] [CrossRef]
- Olmedillas Del Moral, M.; Asavapanumas, N.; Uzcategui, N.L.; Garaschuk, O. Healthy Brain Aging Modifies Microglial Calcium Signaling In Vivo. Int. J. Mol. Sci. 2019, 20, 589. [Google Scholar] [CrossRef] [Green Version]
- Olmedillas Del Moral, M.; Frohlich, N.; Figarella, K.; Mojtahedi, N.; Garaschuk, O. Effect of Caloric Restriction on the in vivo Functional Properties of Aging Microglia. Front. Immunol. 2020, 11, 750. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Sun, W.; Suzuki, K.; Toptunov, D.; Stoyanov, S.; Yuzaki, M.; Khiroug, L.; Dityatev, A. In vivo Two-Photon Imaging of Anesthesia-Specific Alterations in Microglial Surveillance and Photodamage-Directed Motility in Mouse Cortex. Front. Neurosci. 2019, 13, 421. [Google Scholar] [CrossRef] [Green Version]
- Garaschuk, O. Imaging microcircuit function in healthy and diseased brain. Exp. Neurol. 2013, 242, 41–49. [Google Scholar] [CrossRef]
- Riester, K.; Brawek, B.; Savitska, D.; Frohlich, N.; Zirdum, E.; Mojtahedi, N.; Heneka, M.T.; Garaschuk, O. In vivo characterization of functional states of cortical microglia during peripheral inflammation. Brain Behav. Immun. 2020, 87, 243–255. [Google Scholar] [CrossRef]
- Garaschuk, O. The role of NLRP3 inflammasome for microglial response to peripheral inflammation. Neural Regen Res. 2021, 16, 294–295. [Google Scholar] [CrossRef]
- Inoue, K. Purinergic systems in microglia. Cell Mol. Life Sci. 2008, 65, 3074–3080. [Google Scholar] [CrossRef]
- Pozner, A.; Xu, B.; Palumbos, S.; Gee, J.M.; Tvrdik, P.; Capecchi, M.R. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Front. Mol. Neurosci. 2015, 8, 12. [Google Scholar] [CrossRef]
- Brawek, B.; Schwendele, B.; Riester, K.; Kohsaka, S.; Lerdkrai, C.; Liang, Y.; Garaschuk, O. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 2014, 127, 495–505. [Google Scholar] [CrossRef]
- Srinivasan, R.; Huang, B.S.; Venugopal, S.; Johnston, A.D.; Chai, H.; Zeng, H.; Golshani, P.; Khakh, B.S. Ca(2+) signaling in astrocytes from Ip3r2(-/-) mice in brain slices and during startle responses in vivo. Nat. Neurosci. 2015, 18, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Brawek, B.; Liang, Y.; Savitska, D.; Li, K.; Fomin-Thunemann, N.; Kovalchuk, Y.; Zirdum, E.; Jakobsson, J.; Garaschuk, O. A new approach for ratiometric in vivo calcium imaging of microglia. Sci. Rep. 2017, 7, 6030. [Google Scholar] [CrossRef]
- Nissen, J.C. Microglial Function across the Spectrum of Age and Gender. Int. J. Mol. Sci. 2017, 18, 561. [Google Scholar] [CrossRef]
- Mouton, P.R.; Long, J.M.; Lei, D.L.; Howard, V.; Jucker, M.; Calhoun, M.E.; Ingram, D.K. Age and gender effects on microglia and astrocyte numbers in brains of mice. Brain Res. 2002, 956, 30–35. [Google Scholar] [CrossRef]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B.; et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep. 2018, 24, 2773–2783. [Google Scholar] [CrossRef] [Green Version]
- Thion, M.S.; Low, D.; Silvin, A.; Chen, J.; Grisel, P.; Schulte-Schrepping, J.; Blecher, R.; Ulas, T.; Squarzoni, P.; Hoeffel, G.; et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 2018, 172, 500–516. [Google Scholar] [CrossRef] [Green Version]
- Acaz-Fonseca, E.; Duran, J.C.; Carrero, P.; Garcia-Segura, L.M.; Arevalo, M.A. Sex differences in glia reactivity after cortical brain injury. Glia 2015, 63, 1966–1981. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019, 27, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef]
- Bayliak, M.M.; Sorochynska, O.M.; Kuzniak, O.V.; Gospodaryov, D.V.; Demianchuk, O.I.; Vasylyk, Y.V.; Mosiichuk, N.M.; Storey, K.B.; Garaschuk, O.; Lushchak, V.I. Middle age as a turning point in mouse cerebral cortex energy and redox metabolism: Modulation by every-other-day fasting. Exp. Gerontol. 2020, 111182. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; Neher, J.J.; Suhs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J. Neurochem. 2000, 75, 965–972. [Google Scholar] [CrossRef]
- Ikeda, M.; Tsuno, S.; Sugiyama, T.; Hashimoto, A.; Yamoto, K.; Takeuchi, K.; Kishi, H.; Mizuguchi, H.; Kohsaka, S.I.; Yoshioka, T. Ca(2+) spiking activity caused by the activation of store-operated Ca(2+) channels mediates TNF-alpha release from microglial cells under chronic purinergic stimulation. Biochim. Biophys. Acta 2013, 1833, 2573–2585. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.K.; Kettenmann, H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): Suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar]
- Nikodemova, M.; Small, A.L.; Kimyon, R.S.; Watters, J.J. Age-dependent differences in microglial responses to systemic inflammation are evident as early as middle age. Physiol. Genom. 2016, 48, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J. Neuroinflamm. 2009, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Liu, X.; Queen, N.J.; Patel, R.S.; Wilkins, R.K.; Mo, X.; Cao, L. Long-term environmental enrichment affects microglial morphology in middle age mice. Aging (Albany N. Y.) 2019, 11, 2388–2402. [Google Scholar] [CrossRef]
- McMurphy, T.; Huang, W.; Queen, N.J.; Ali, S.; Widstrom, K.J.; Liu, X.; Xiao, R.; Siu, J.J.; Cao, L. Implementation of environmental enrichment after middle age promotes healthy aging. Aging (Albany N. Y.) 2018, 10, 1698–1721. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Moller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Olah, M.; Patrick, E.; Villani, A.C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020, 68, 845–854. [Google Scholar] [CrossRef]
- Pluvinage, J.V.; Haney, M.S.; Smith, B.A.H.; Sun, J.; Iram, T.; Bonanno, L.; Li, L.; Lee, D.P.; Morgens, D.W.; Yang, A.C.; et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 2019, 568, 187–192. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Moller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.E.; Zettel, M.L.; Ison, J.R.; Allen, P.D.; Majewska, A.K. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 2012, 60, 541–558. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195. [Google Scholar] [CrossRef] [Green Version]
- Floden, A.M.; Combs, C.K. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J. Alzheimers Dis. 2011, 25, 279–293. [Google Scholar] [CrossRef]
- Mangold, C.A.; Wronowski, B.; Du, M.; Masser, D.R.; Hadad, N.; Bixler, G.V.; Brucklacher, R.M.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J. Neuroinflamm. 2017, 14, 141. [Google Scholar] [CrossRef]
- Moretti, M.; Zoli, M.; George, A.A.; Lukas, R.J.; Pistillo, F.; Maskos, U.; Whiteaker, P.; Gotti, C. The novel alpha7beta2-nicotinic acetylcholine receptor subtype is expressed in mouse and human basal forebrain: Biochemical and pharmacological characterization. Mol. Pharmacol. 2014, 86, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I.; Moretti, M.; Pedrazzi, P.; Pucci, L.; Zoli, M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lu, Y.; Bian, H.; Guo, L.; Zhu, H. Activation of the alpha7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am. J. Transl. Res. 2017, 9, 971–985. [Google Scholar]
- Wang, Y.; Zhu, N.; Wang, K.; Zhang, Z.; Wang, Y. Identification of alpha7 nicotinic acetylcholine receptor on hippocampal astrocytes cultured in vitro and its role on inflammatory mediator secretion. Neural Regen. Res. 2012, 7, 1709–1714. [Google Scholar] [CrossRef]
- Dickinson, J.A.; Kew, J.N.; Wonnacott, S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol. Pharmacol. 2008, 74, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Resende, R.R.; Adhikari, A. Cholinergic receptor pathways involved in apoptosis, cell proliferation and neuronal differentiation. Cell Commun. Signal. 2009, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, Z.; Jiang, Y.Y.; Shen, W.X.; Peng, Y.P.; Qiu, Y.H. Acetylcholine suppresses microglial inflammatory response via alpha7nAChR to protect hippocampal neurons. J. Integr. Neurosci. 2019, 18, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Noda, M.; Kobayashi, A.I. Nicotine inhibits activation of microglial proton currents via interactions with alpha7 acetylcholine receptors. J. Physiol. Sci. 2017, 67, 235–245. [Google Scholar] [CrossRef]
- Cortes, M.; Cao, M.; Liu, H.L.; Moore, C.S.; Durosier, L.D.; Burns, P.; Fecteau, G.; Desrochers, A.; Barreiro, L.B.; Antel, J.P.; et al. alpha7 nicotinic acetylcholine receptor signaling modulates the inflammatory phenotype of fetal brain microglia: First evidence of interference by iron homeostasis. Sci. Rep. 2017, 7, 10645. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, C.; Jiang, L.; Li, M.; Long, T.; He, W.; Qin, G.; Chen, L.; Zhou, J. alpha7 Nicotinic acetylcholine receptor-mediated anti-inflammatory effect in a chronic migraine rat model via the attenuation of glial cell activation. J. Pain Res. 2018, 11, 1129–1140. [Google Scholar] [CrossRef] [Green Version]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; Leon, R.; Lopez, M.G. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef]
- Xue, R.; Wan, Y.; Sun, X.; Zhang, X.; Gao, W.; Wu, W. Nicotinic Mitigation of Neuroinflammation and Oxidative Stress After Chronic Sleep Deprivation. Front. Immunol. 2019, 10, 2546. [Google Scholar] [CrossRef]
- Toman, J.; Fiskum, G. Influence of aging on membrane permeability transition in brain mitochondria. J. Bioenerg. Biomembr. 2011, 43, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Navarro, E.; Gonzalez-Lafuente, L.; Perez-Liebana, I.; Buendia, I.; Lopez-Bernardo, E.; Sanchez-Ramos, C.; Prieto, I.; Cuadrado, A.; Satrustegui, J.; Cadenas, S.; et al. Heme-Oxygenase I and PCG-1alpha Regulate Mitochondrial Biogenesis via Microglial Activation of Alpha7 Nicotinic Acetylcholine Receptors Using PNU282987. Antioxid. Redox Signal. 2017, 27, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Skok, M.; Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Uspenska, K. Nicotinic acetylcholine receptors in mitochondria: Subunit composition, function and signalling. Neurotransmitter 2016, 3, e1290. [Google Scholar] [CrossRef]
- Kalashnyk, O.; Lykhmus, O.; Uspenska, K.; Izmailov, M.; Komisarenko, S.; Skok, M. Mitochondrial alpha7 nicotinic acetylcholine receptors are displaced from complexes with VDAC1 to form complexes with Bax upon apoptosis induction. Int. J. Biochem. Cell Biol. 2020, 129, 105879. [Google Scholar] [CrossRef]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express alpha7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: Study on isolated mitochondria. PLoS ONE 2012, 7, e31361. [Google Scholar] [CrossRef]
- Uspenska, K.; Lykhmus, O.; Arias, H.R.; Pons, S.; Maskos, U.; Komisarenko, S.; Skok, M. Positive allosteric modulators of alpha7* or beta2* nicotinic acetylcholine receptors trigger different kinase pathways in mitochondria. Int. J. Biochem. Cell Biol. 2018, 99, 226–235. [Google Scholar] [CrossRef]
- Lykhmus, O.; Voytenko, L.; Koval, L.; Mykhalskiy, S.; Kholin, V.; Peschana, K.; Zouridakis, M.; Tzartos, S.; Komisarenko, S.; Skok, M. alpha7 Nicotinic acetylcholine receptor-specific antibody induces inflammation and amyloid beta42 accumulation in the mouse brain to impair memory. PLoS ONE 2015, 10, e0122706. [Google Scholar] [CrossRef]
- Krestinina, O.; Azarashvili, T.; Baburina, Y.; Galvita, A.; Grachev, D.; Stricker, R.; Reiser, G. In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2’,3’-cyclic nucleotide-3’-phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 2015, 80, 41–50. [Google Scholar] [CrossRef]
- Shao, B.Z.; Ke, P.; Xu, Z.Q.; Wei, W.; Cheng, M.H.; Han, B.Z.; Chen, X.W.; Su, D.F.; Liu, C. Autophagy Plays an Important Role in Anti-inflammatory Mechanisms Stimulated by Alpha7 Nicotinic Acetylcholine Receptor. Front. Immunol. 2017, 8, 553. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, S.R.; Choi, S.S.; Yeo, H.G.; Chang, K.T.; Lee, H.J. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed. Res. Int. 2014, 2014, 297241. [Google Scholar] [CrossRef]
- Guan, Y.Z.; Jin, X.D.; Guan, L.X.; Yan, H.C.; Wang, P.; Gong, Z.; Li, S.J.; Cao, X.; Xing, Y.L.; Gao, T.M. Nicotine inhibits microglial proliferation and is neuroprotective in global ischemia rats. Mol. Neurobiol. 2015, 51, 1480–1488. [Google Scholar] [CrossRef]
- Han, Z.; Shen, F.; He, Y.; Degos, V.; Camus, M.; Maze, M.; Young, W.L.; Su, H. Activation of alpha-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. PLoS ONE 2014, 9, e105711. [Google Scholar] [CrossRef]
- Ellis, A.; Bennett, D.L. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 2013, 111, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Pickering, G.; Marcoux, M.; Chapiro, S.; David, L.; Rat, P.; Michel, M.; Bertrand, I.; Voute, M.; Wary, B. An Algorithm for Neuropathic Pain Management in Older People. Drugs Aging 2016, 33, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Chen, Y.; Wei, H.; Feng, H.; Chang, R.; Yu, D.; Wang, X.; Gong, X.; Zhang, M. Activation of alpha7 acetylcholine receptors reduces neuropathic pain by decreasing dynorphin A release from microglia. Brain Res. 2019, 1715, 57–65. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Lykhmus, O.; Gergalova, G.; Zouridakis, M.; Tzartos, S.; Komisarenko, S.; Skok, M. Inflammation decreases the level of alpha7 nicotinic acetylcholine receptors in the brain mitochondria and makes them more susceptible to apoptosis induction. Int. Immunopharmacol. 2015, 29, 148–151. [Google Scholar] [CrossRef]
- Alkondon, M.; Albuquerque, E.X. Subtype-specific inhibition of nicotinic acetylcholine receptors by choline: A regulatory pathway. J. Pharmacol. Exp. Ther. 2006, 318, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Danne, O.; Mockel, M. Choline in acute coronary syndrome: An emerging biomarker with implications for the integrated assessment of plaque vulnerability. Expert. Rev. Mol. Diagn. 2010, 10, 159–171. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef] [Green Version]
- Shimohama, S.; Kawamata, J. Roles of Nicotinic Acetylcholine Receptors in the Pathology and Treatment of Alzheimer’s and Parkinson’s Diseases. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Akaike, A., Shimohama, S., Misu, Y., Eds.; Springer: Singapore, 2018; pp. 137–158. [Google Scholar] [CrossRef] [Green Version]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Abeta accumulation through suppression of neuronal gamma-secretase activity and promotion of microglial amyloid-beta phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar] [CrossRef]
- Suzuki, S.; Kawamata, J.; Matsushita, T.; Matsumura, A.; Hisahara, S.; Takata, K.; Kitamura, Y.; Kem, W.; Shimohama, S. 3-[(2,4-Dimethoxy)benzylidene]-anabaseine dihydrochloride protects against 6-hydroxydopamine-induced parkinsonian neurodegeneration through alpha7 nicotinic acetylcholine receptor stimulation in rats. J. Neurosci. Res. 2013, 91, 462–471. [Google Scholar] [CrossRef]
- Marks, S.M.; Lockhart, S.N.; Baker, S.L.; Jagust, W.J. Tau and beta-Amyloid Are Associated with Medial Temporal Lobe Structure, Function, and Memory Encoding in Normal Aging. J. Neurosci. 2017, 37, 3192–3201. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, K.; Jiang, D.; Wang, Y.; Xiao, X.; Zhu, N.; Li, M.; Jia, S.; Wang, Y. Blocking mPTP on Neural Stem Cells and Activating the Nicotinic Acetylcholine Receptor alpha7 Subunit on Microglia Attenuate Abeta-Induced Neurotoxicity on Neural Stem Cells. Neurochem. Res. 2016, 41, 1483–1495. [Google Scholar] [CrossRef]
- Foucault-Fruchard, L.; Antier, D. Therapeutic potential of alpha7 nicotinic receptor agonists to regulate neuroinflammation in neurodegenerative diseases. Neural Regen. Res. 2017, 12, 1418–1421. [Google Scholar] [CrossRef]
- Papke, R.L. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 2014, 89, 1–11. [Google Scholar] [CrossRef] [Green Version]
- King, J.R.; Gillevet, T.C.; Kabbani, N. A G protein-coupled alpha7 nicotinic receptor regulates signaling and TNF-alpha release in microglia. FEBS Open Bio 2017, 7, 1350–1361. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Mikkelsen, J.D. The alpha7 nicotinic acetylcholine receptor ligands methyllycaconitine, NS6740 and GTS-21 reduce lipopolysaccharide-induced TNF-alpha release from microglia. J. Neuroimmunol. 2012, 251, 65–72. [Google Scholar] [CrossRef]
- Steiner, L.; Gold, M.; Mengel, D.; Dodel, R.; Bach, J.P. The endogenous alpha7 nicotinic acetylcholine receptor antagonist kynurenic acid modulates amyloid-beta-induced inflammation in BV-2 microglial cells. J. Neurol. Sci. 2014, 344, 94–99. [Google Scholar] [CrossRef]
- Kalappa, B.I.; Sun, F.; Johnson, S.R.; Jin, K.; Uteshev, V.V. A positive allosteric modulator of alpha7 nAChRs augments neuroprotective effects of endogenous nicotinic agonists in cerebral ischaemia. Br. J. Pharmacol. 2013, 169, 1862–1878. [Google Scholar] [CrossRef] [Green Version]
- Alzarea, S.; Rahman, S. Alpha-7 nicotinic receptor allosteric modulator PNU120596 prevents lipopolysaccharide-induced anxiety, cognitive deficit and depression-like behaviors in mice. Behav. Brain Res. 2019, 366, 19–28. [Google Scholar] [CrossRef]
- Lykhmus, O.; Kalashnyk, O.; Uspenska, K.; Skok, M. Positive Allosteric Modulation of Alpha7 Nicotinic Acetylcholine Receptors Transiently Improves Memory but Aggravates Inflammation in LPS-Treated Mice. Front. Aging Neurosci. 2019, 11, 359. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Bok, E.; Jo, M.; Lee, S.; Lee, B.R.; Kim, J.; Kim, H.J. Dietary Restriction and Neuroinflammation: A Potential Mechanistic Link. Int. J. Mol. Sci 2019, 20, 464. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.; Aon, M.; Gonzalez-Reyes, J.A.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B.; et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef] [Green Version]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and Stress. Neuropsychopharmacology 2017, 42, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Weindruch, R.; Prolla, T.A. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000, 25, 294–297. [Google Scholar] [CrossRef]
- Wahl, D.; Solon-Biet, S.M.; Wang, Q.P.; Wali, J.A.; Pulpitel, T.; Clark, X.; Raubenheimer, D.; Senior, A.M.; Sinclair, D.A.; Cooney, G.J.; et al. Comparing the Effects of Low-Protein and High-Carbohydrate Diets and Caloric Restriction on Brain Aging in Mice. Cell Rep. 2018, 25, 2234–2243. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Raj, D.D.; Schaafsma, W.; van der Heijden, R.A.; Kooistra, S.M.; Reijne, A.C.; Zhang, X.; Moser, J.; Brouwer, N.; Heeringa, P.; et al. Low-Fat Diet With Caloric Restriction Reduces White Matter Microglia Activation During Aging. Front. Mol. Neurosci. 2018, 11, 65. [Google Scholar] [CrossRef]
- Wong, A.M.; Patel, N.V.; Patel, N.K.; Wei, M.; Morgan, T.E.; de Beer, M.C.; de Villiers, W.J.; Finch, C.E. Macrosialin increases during normal brain aging are attenuated by caloric restriction. Neurosci. Lett. 2005, 390, 76–80. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Exercise, microglia, and beyond—workout to communicate with microglia. Neural Regen. Res. 2020, 15, 2029–2030. [Google Scholar] [CrossRef]
- Mee-Inta, O.; Zhao, Z.W.; Kuo, Y.M. Physical Exercise Inhibits Inflammation and Microglial Activation. Cells 2019, 8, 691. [Google Scholar] [CrossRef] [Green Version]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. eLife 2016, 5. [Google Scholar] [CrossRef]
- Giorgetti, E.; Panesar, M.; Zhang, Y.; Joller, S.; Ronco, M.; Obrecht, M.; Lambert, C.; Accart, N.; Beckmann, N.; Doelemeyer, A.; et al. Modulation of Microglia by Voluntary Exercise or CSF1R Inhibition Prevents Age-Related Loss of Functional Motor Units. Cell Rep. 2019, 29, 1539–1554. [Google Scholar] [CrossRef]
- Sung, Y.H.; Kim, S.C.; Hong, H.P.; Park, C.Y.; Shin, M.S.; Kim, C.J.; Seo, J.H.; Kim, D.Y.; Kim, D.J.; Cho, H.J. Treadmill exercise ameliorates dopaminergic neuronal loss through suppressing microglial activation in Parkinson’s disease mice. Life Sci. 2012, 91, 1309–1316. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brawek, B.; Skok, M.; Garaschuk, O. Changing Functional Signatures of Microglia along the Axis of Brain Aging. Int. J. Mol. Sci. 2021, 22, 1091. https://doi.org/10.3390/ijms22031091
Brawek B, Skok M, Garaschuk O. Changing Functional Signatures of Microglia along the Axis of Brain Aging. International Journal of Molecular Sciences. 2021; 22(3):1091. https://doi.org/10.3390/ijms22031091
Chicago/Turabian StyleBrawek, Bianca, Maryna Skok, and Olga Garaschuk. 2021. "Changing Functional Signatures of Microglia along the Axis of Brain Aging" International Journal of Molecular Sciences 22, no. 3: 1091. https://doi.org/10.3390/ijms22031091
APA StyleBrawek, B., Skok, M., & Garaschuk, O. (2021). Changing Functional Signatures of Microglia along the Axis of Brain Aging. International Journal of Molecular Sciences, 22(3), 1091. https://doi.org/10.3390/ijms22031091