
Glutamine Uptake via SNAT6 and Caveolin Regulates Glutamine–Glutamate Cycle

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Structural Representation of SNAT6 Protein with TMS Prediction and 3D Modeling
2.2. Several Genes Relevant to Glutamate–Glutamine Cycle Are Predicted to Interact with SNAT6
2.3. siRNA-Induced Knockdown of SNAT6 Shows Upregulation or Downregulation of Predicted Interacting Genes
2.4. CTPs2 Shows Similar Histological Profile as SNAT6
2.5. Grm2 Is Co-Expressed with both SNAT6 and CTPs2
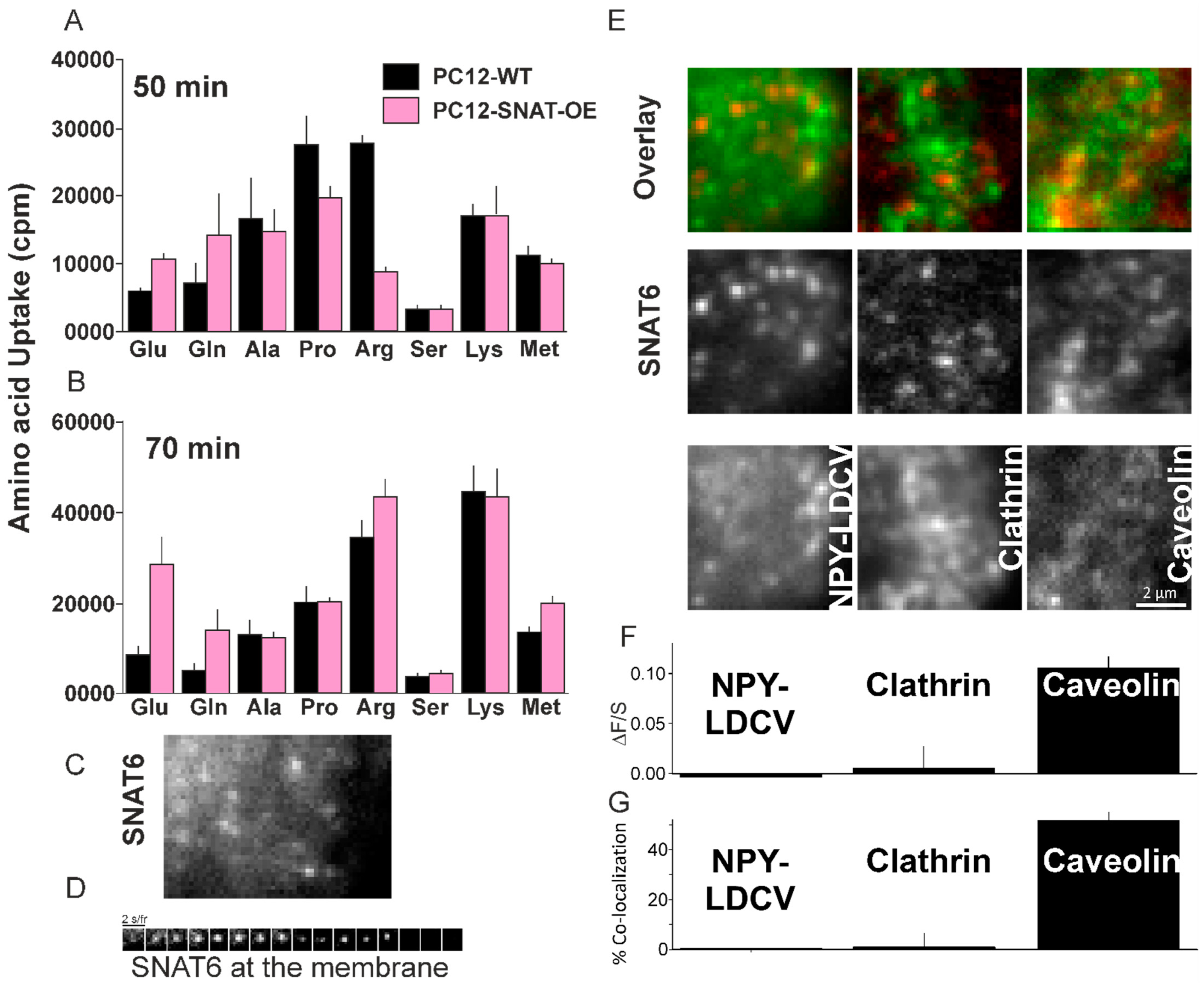
2.6. Glutamine and Glutamate Are Preferred Substrate for SNAT6
2.7. SNAT6 Localizes with Caveolin1
2.8. SNAT6 Associated Caveolin Complexes Internalize in Response to Glutamine and Glutamate
2.9. SNAT6 Associated Caveolin Internalization Is Dependent on Availability of Na+
2.10. SNAT6 Alocalization and Downstream Signaling
3. Discussion
4. Methods
4.1. Sequence and Homology Modeling
4.2. Microarray Data and ARACNE Analysis for SNAT6
4.3. Cell Cultures and Cell Lines
4.4. Knockdown of SNAT6
4.5. Quantitative Real-Time PCR (qPCR) and Data Analysis
- (1)
- 2-DDCt method was used [39], where the differences in the cycle threshold (Ct) values between the house keeping gene and a target gene, with or without treatment, was calculated. Then, the difference between these values was calculated as follows: Ct(treated) − Ct(non-treated) = (Ct(gene)-housekeeping) treated − (Ct(gene)-housekeeping) non-treated. To determine the ratio of expression levels in treated sample versus non-treated sample, the Qr formula was used as follows: Qr = 2 − Ct(treated) − Ct(non-treated).
- (2)
- Efficiency-corrected Pfaffl Method was then performed. The fold difference is given by 1.85(A − B)/1.97(F − G) where A = average Cq of target gene in non-treated sample, B = average Cq of target gene in treated sample, F = average Cq of housekeeping reference gene in non-treated sample and G = average Cq of reference gene in treated sample. Primer efficiency value of the target gene is 1.85 while that of the the reference gene is 1.97 (both were computed according to LinReg).
- (3)
- The third equation used the same equation as in two but instead of using the mean value, the minimum value was used to retrieve the lowest mean cycle threshold and then all quantities for this particular gene was expressed relative to this reaction. Finally, the graph was made using software GraphPad Prism 5.
4.6. Tissue Collection and Sectioning
4.7. Fluorescent Immunohistochemistry on Paraffin Embedded Sections
4.8. Plasmid Constructs
4.9. Proximity Ligation Assay (PLA)
4.10. TIRF Microscopy
4.11. Image Analysis
4.12. Uptake Assays Using Tritium Labeled Amino Acids
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almén, M.S.; Nordström, K.J.V.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perland, E.; Fredriksson, R. Classification Systems of Secondary Active Transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Fredriksson, R.; Schiöth, H.B. Solute carriers as drug targets: Current use, clinical trials and prospective. Mol. Asp. Med. 2013, 34, 702–710. [Google Scholar] [CrossRef] [PubMed]
- César-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.F.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Willard, S.S.; Koochekpour, S. Glutamate Signaling in Benign and Malignant Disorders: Current Status, Future Perspectives, and Therapeutic Implications. Int. J. Biol. Sci. 2013, 9, 728–742. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.D. Functional identification of activity-regulated, high-affinity glutamine transport in hippocampal neurons inhibited by riluzole. J. Neurochem. 2017, 142, 29–40. [Google Scholar] [CrossRef]
- Lichter-Konecki, U. Profiling of astrocyte properties in the hyperammonaemic brain: Shedding new light on the pathophysiology of the brain damage in hyperammonaemia. J. Inherit. Metab. Dis. 2008, 31, 492–502. [Google Scholar] [CrossRef]
- Albrecht, J.; Sidoryk-Węgrzynowicz, M.; Zielińska, M.; Aschner, M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010, 6, 263–276. [Google Scholar] [CrossRef]
- Bagchi, S.; Baomar, H.A.; Al-Walai, S.; Al-Sadi, S.; Fredriksson, R. Histological Analysis of SLC38A6 (SNAT6) Expression in Mouse Brain Shows Selective Expression in Excitatory Neurons with High Expression in the Synapses. PLoS ONE 2014, 9, e95438. [Google Scholar] [CrossRef] [Green Version]
- Todd, A.C.; Marx, M.-C.; Hulme, S.R.; Bröer, S.; Billups, B. SNAT3-mediated glutamine transport in perisynaptic astrocytesin situis regulated by intracellular sodium. GLIA 2017, 65, 900–916. [Google Scholar] [CrossRef]
- González, M.I.; Krizman-Genda, E.; Robinson, M.B. Caveolin-1 Regulates the Delivery and Endocytosis of the Glutamate Transporter, Excitatory Amino Acid Carrier 1. J. Biol. Chem. 2007, 282, 29855–29865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bröer, S. The SLC38 family of sodium–amino acid co-transporters. Pflüg. Archiv Eur. J. Physiol. 2013, 466, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.D.; Hare, N.; Bubb, W.A.; McEwan, S.R.; Bröer, A.; McQuillan, J.A.; Balcar, V.J.; Conigrave, A.D.; Bröer, S. Inhibition of glutamine transport depletes glutamate and GABA neurotransmitter pools: Further evidence for metabolic compartmentation. J. Neurochem. 2003, 85, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, F.; Melone, M. The glutamine commute: Lost in the tube? Neurochem. Int. 2006, 48, 459–464. [Google Scholar] [CrossRef]
- Bansal, M.; Belcastro, V.; Ambesi-Impiombato, A.; Di Bernardo, D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla-Favera, R.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Van Someren, E.; Wessels, L.; Backer, E.; Reinders, M. Genetic network modeling. Pharmacogenomics 2002, 3, 507–525. [Google Scholar] [CrossRef]
- Hellsten, S.V.; Lekholm, E.; Ahmad, T.; Fredriksson, R. The gene expression of numerous SLC transporters is altered in the immortalized hypothalamic cell line N25/2 following amino acid starvation. FEBS Open Bio 2017, 7, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Butchbach, M.E.; Tian, G.; Guo, H.; Lin, C.-L.G.; Tzameli, I.; Fang, H.; Ollero, M.; Shi, H.; Hamm, J.K.; Kievit, P.; et al. Association of Excitatory Amino Acid Transporters, Especially EAAT2, with Cholesterol-rich Lipid Raft Microdomains. J. Biol. Chem. 2004, 279, 34388–34396. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Royal, C.; Dupuis, J.P.; Varela, J.A.; Panatier, A.; Pinson, B.; Baufreton, J.; Groc, L.; Oliet, S.H.R. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 2015, 18, 219–226. [Google Scholar] [CrossRef]
- Head, B.P.; Insel, P.A. Do caveolins regulate cells by actions outside of caveolae? Trends Cell Biol. 2007, 17, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2005, 22, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef]
- Hellsten, S.V.; Hägglund, M.G.; Eriksson, M.M.; Fredriksson, R. The neuronal and astrocytic protein SLC38A10 transports glutamine, glutamate, and aspartate, suggesting a role in neurotransmission. FEBS Open Bio 2017, 7, 730–746. [Google Scholar] [CrossRef] [Green Version]
- Gandasi, N.; Barg, S. Contact-induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site. Nat. Commun. 2014, 5, 3914. [Google Scholar] [CrossRef] [Green Version]
- Schiöth, H.B.; Roshanbin, S.; Hägglund, M.G.; Fredriksson, R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol. Asp. Med. 2013, 34, 571–585. [Google Scholar] [CrossRef]
- Omar-Hmeadi, M.; Gandasi, N.; Barg, S. PtdIns(4,5)P2 is not required for secretory granule docking. Traffic 2018, 19, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Nguyen, P.M.; Guček, A.; Thonig, A.; Barg, S.; Idevall-Hagren, O. Plasma Membrane Phosphatidylinositol 4,5-Bisphosphate Regulates Ca2+ -Influx and Insulin Secretion from Pancreatic β Cells. Cell Chem. Biol. 2016, 23, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced changes in protein compared to mRNA levels across non-proliferating tissues. BMC Genom. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Samuvel, D.J.; Jayanthi, L.D.; Bhat, N.R.; Ramamoorthy, S. A Role for p38 Mitogen-Activated Protein Kinase in the Regulation of the Serotonin Transporter: Evidence for Distinct Cellular Mechanisms Involved in Transporter Surface Expression. J. Neurosci. 2005, 25, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Fecchi, K.; Volonte, D.; Hezel, M.P.; Schmeck, K.; Galbiati, F. Spatial and temporal regulation of GLUT4 translocation by flotillin-1 and caveolin-3 in skeletal muscle cells. FASEB J. 2006, 20, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Underhill, S.M.; Wheeler, D.S.; Li, M.; Watts, S.D.; Ingram, S.L.; Amara, S.G. Amphetamine Modulates Excitatory Neurotransmission through Endocytosis of the Glutamate Transporter EAAT3 in Dopamine Neurons. Neuron 2014, 83, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Menchini, R.J.; Chaudhry, F.A. Multifaceted regulation of the system A transporter Slc38a2 suggests nanoscale regulation of amino acid metabolism and cellular signaling. Neuropharmacology 2019, 161, 107789. [Google Scholar] [CrossRef]
- Alshammari, M.A.; Alshammari, T.K.; Laezza, F. Improved Methods for Fluorescence Microscopy Detection of Macromolecules at the Axon Initial Segment. Front. Cell. Neurosci. 2016, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Lamberts, R.; Goldsmith, P.C. Fixation, fine structure, and immunostaining for neuropeptides: Perfusion versus immersion of the neuroendocrine hypothalamus. J. Histochem. Cytochem. 1986, 34, 389–398. [Google Scholar] [CrossRef]
- Tawfik, D.; Zaccagnino, A.; Bernt, A.; Szczepanowski, M.; Klapper, W.; Schwab, A.; Kalthoff, H.; Trauzold, A. The A818–6 system as an in-vitro model for studying the role of the transportome in pancreatic cancer. BMC Cancer 2020, 20, 1–17. [Google Scholar] [CrossRef]
- Ye, J.; Xu, B.; Fan, B.; Zhang, J.; Yuan, F.; Chen, Y.; Sun, Z.; Yan, X.; Song, Y.; Song, S.; et al. Discovery of Selenocysteine as a Potential Nanomedicine Promotes Cartilage Regeneration with Enhanced Immune Response by Text Mining and Biomedical Databases. Front. Pharmacol. 2020, 11, 1138. [Google Scholar] [CrossRef]
- Grainger, A.T.; Jones, M.B.; Chen, M.-H.; Shi, W. Polygenic Control of Carotid Atherosclerosis in a BALB/cJ × SM/J Intercross and a Combined Cross Involving Multiple Mouse Strains. G3 Genes Genomes Genet. 2017, 7, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Li, C. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 2001, 98, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Ai, H.-W.; Wong, P.; Young, J.D.; Campbell, R.E.; Casey, J.R. Red Fluorescent Protein pH Biosensor to Detect Concentrative Nucleoside Transport. J. Biol. Chem. 2009, 284, 20499–20511. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Rihani, A.; Van Goethem, A.; Ongenaert, M.; De Brouwer, S.; Volders, P.-J.; Agarwal, S.; De Preter, K.; Mestdagh, P.; Shohet, J.; Speleman, F.; et al. Genome wide expression profiling of p53 regulated miRNAs in neuroblastoma. Sci. Rep. 2015, 5, srep09027. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.J.; Goergen, P.; Rajendran, J.; Zheleznyakova, G.Y.; Hägglund, M.G.; Perland, E.; Bagchi, S.; Kalogeropoulou, A.; Khan, Z.; Fredriksson, R.; et al. Obesity-Linked Homologues TfAP-2 and Twz Establish Meal Frequency in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004499. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Taylor, M.J.; Perrais, D.; Merrifield, C.J. A High Precision Survey of the Molecular Dynamics of Mammalian Clathrin-Mediated Endocytosis. PLoS Biol. 2011, 9, e1000604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandasi, N.; Vestö, K.; Helou, M.; Yin, P.; Saras, J.; Barg, S. Survey of Red Fluorescence Proteins as Markers for Secretory Granule Exocytosis. PLoS ONE 2015, 10, e0127801. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, T.P.; Ahn, S.; Meyer, T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 1998, 8, 343–346. [Google Scholar] [CrossRef] [Green Version]
- Jarvius, M.; Paulsson, J.; Weibrecht, I.; Leuchowius, K.-J.; Andersson, A.-C.; Wählby, C.; Gullberg, M.; Botling, J.; Sjöblom, T.; Markova, B.; et al. In SituDetection of Phosphorylated Platelet-derived Growth Factor Receptor β Using a Generalized Proximity Ligation Method. Mol. Cell. Proteom. 2007, 6, 1500–1509. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstråle, K.; Leuchowius, K.-J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.-G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef]
- Gullberg, M.; Gústafsdóttir, S.M.; Schallmeiner, E.; Jarvius, J.; Bjarnegård, M.; Betsholtz, C.; Landegren, U.; Fredriksson, S. Cytokine detection by antibody-based proximity ligation. Proc. Natl. Acad. Sci. USA. 2004, 101, 8420–8424. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Östman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Taraska, J.W.; Perrais, D.; Ohara-Imaizumi, M.; Nagamatsu, S.; Almers, W. Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.-A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpen-ter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandasi, N.R.; Arapi, V.; Mickael, M.E.; Belekar, P.A.; Granlund, L.; Kothegala, L.; Fredriksson, R.; Bagchi, S. Glutamine Uptake via SNAT6 and Caveolin Regulates Glutamine–Glutamate Cycle. Int. J. Mol. Sci. 2021, 22, 1167. https://doi.org/10.3390/ijms22031167
Gandasi NR, Arapi V, Mickael ME, Belekar PA, Granlund L, Kothegala L, Fredriksson R, Bagchi S. Glutamine Uptake via SNAT6 and Caveolin Regulates Glutamine–Glutamate Cycle. International Journal of Molecular Sciences. 2021; 22(3):1167. https://doi.org/10.3390/ijms22031167
Chicago/Turabian StyleGandasi, Nikhil R., Vasiliki Arapi, Michel E. Mickael, Prajakta A. Belekar, Louise Granlund, Lakshmi Kothegala, Robert Fredriksson, and Sonchita Bagchi. 2021. "Glutamine Uptake via SNAT6 and Caveolin Regulates Glutamine–Glutamate Cycle" International Journal of Molecular Sciences 22, no. 3: 1167. https://doi.org/10.3390/ijms22031167
APA StyleGandasi, N. R., Arapi, V., Mickael, M. E., Belekar, P. A., Granlund, L., Kothegala, L., Fredriksson, R., & Bagchi, S. (2021). Glutamine Uptake via SNAT6 and Caveolin Regulates Glutamine–Glutamate Cycle. International Journal of Molecular Sciences, 22(3), 1167. https://doi.org/10.3390/ijms22031167