
Gene Editing Correction of a Urea Cycle Defect in Organoid Stem Cell Derived Hepatocyte-like Cells

 ,
, 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
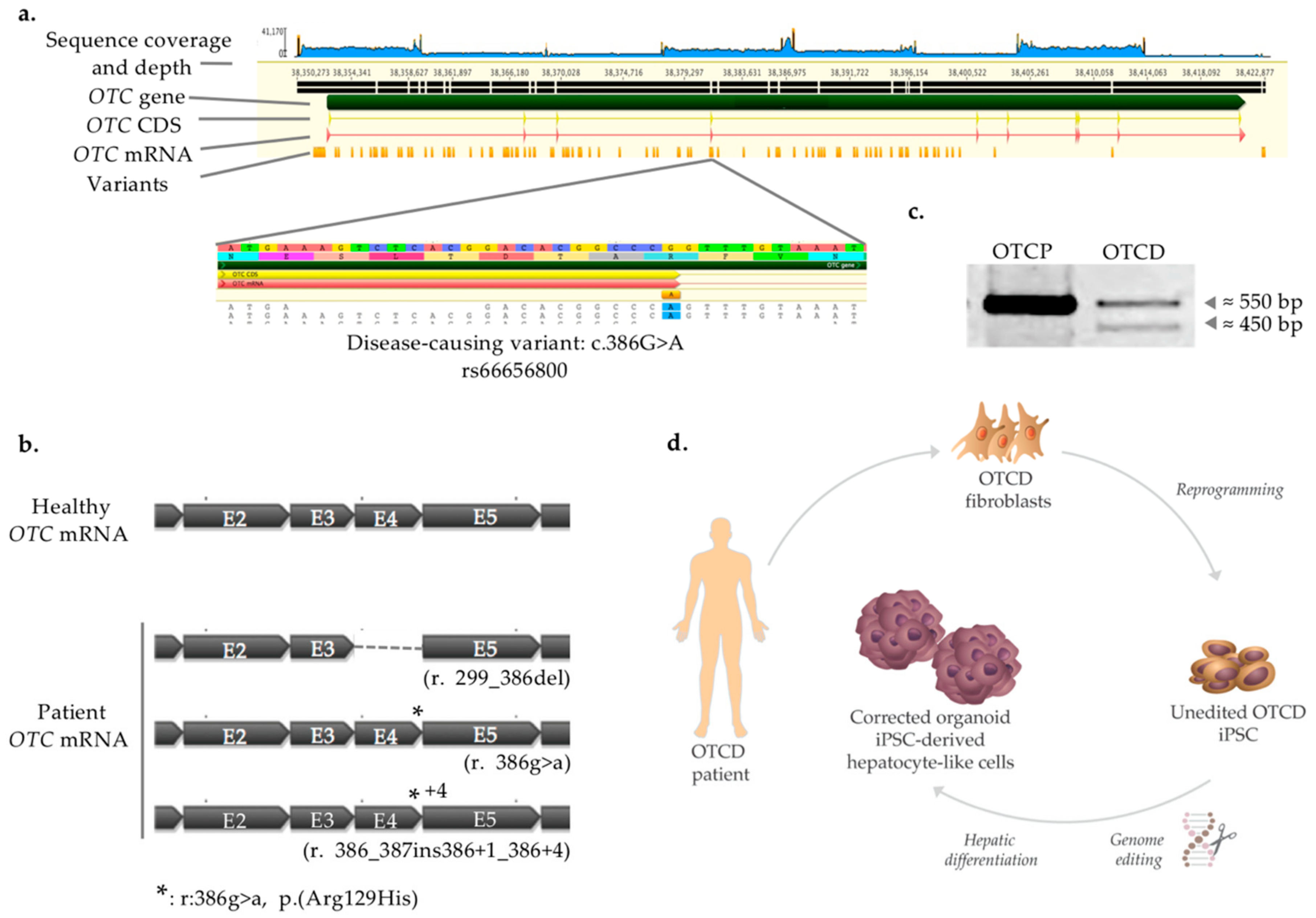
2.1. Disease-Causing Variant Identification and Transcript Characterization
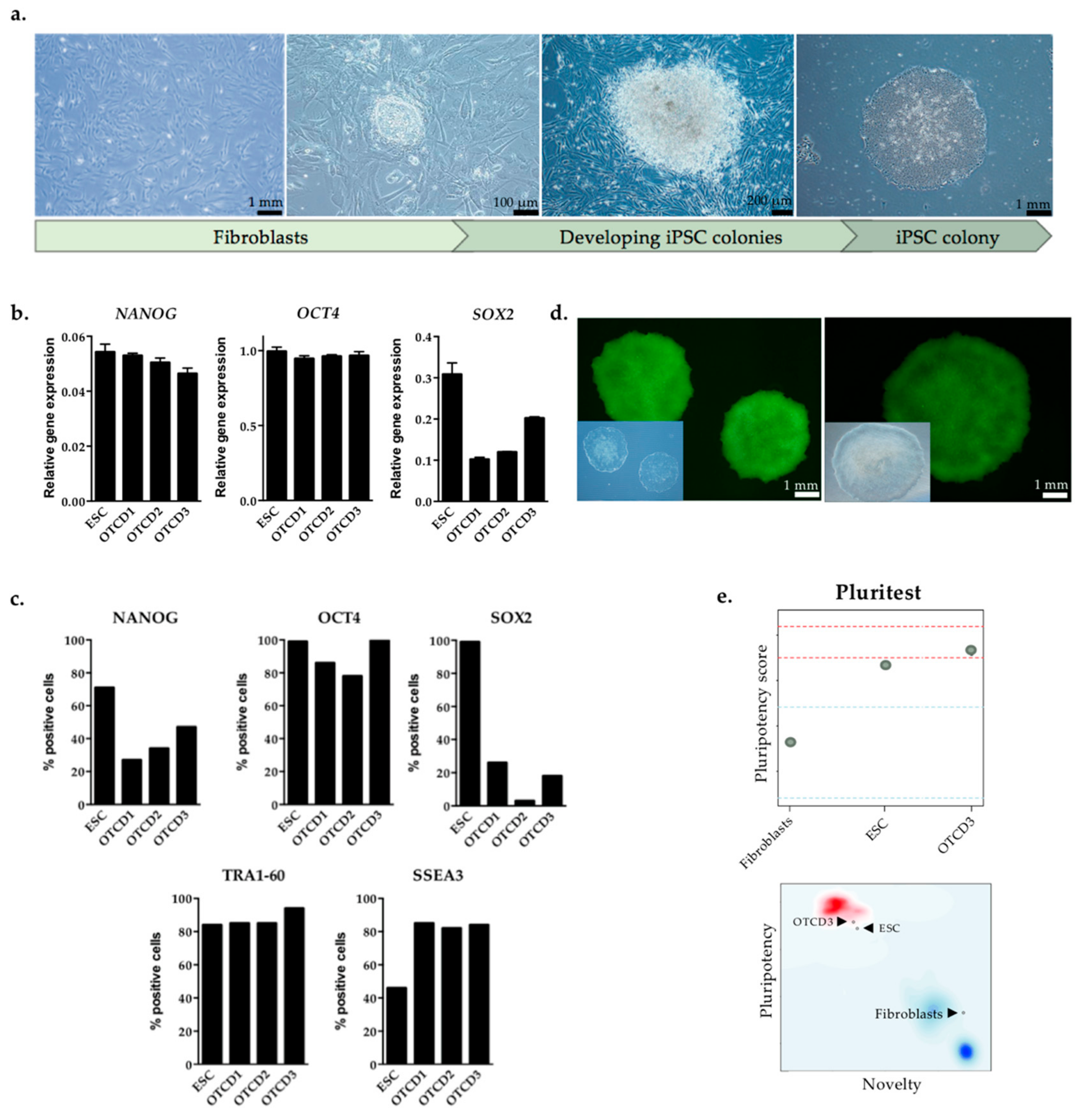
2.2. Generation and Characterization of Patient-Derived iPSC
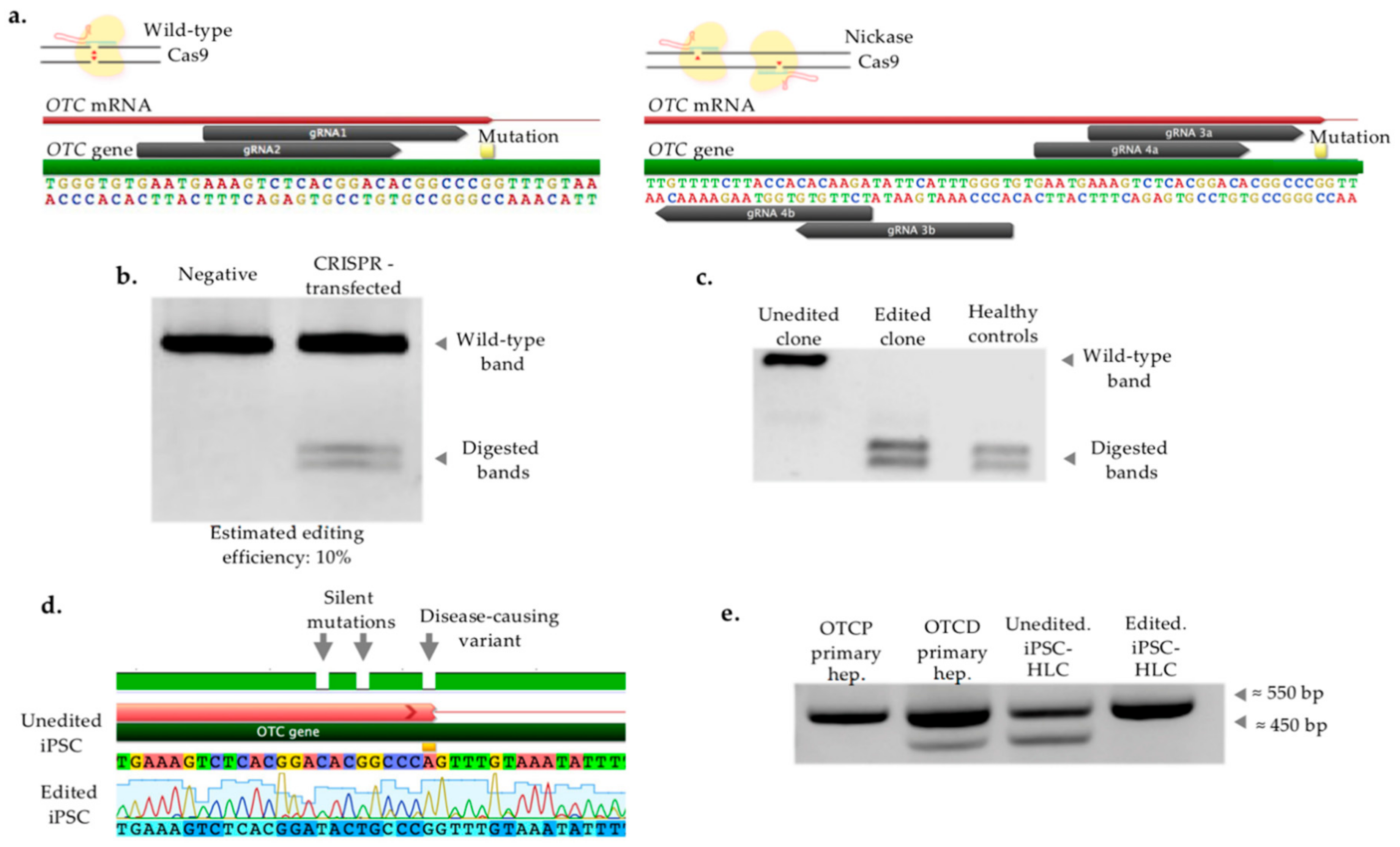
2.3. CRISPR/Cas-Mediated Genome Editing of Patient iPSC
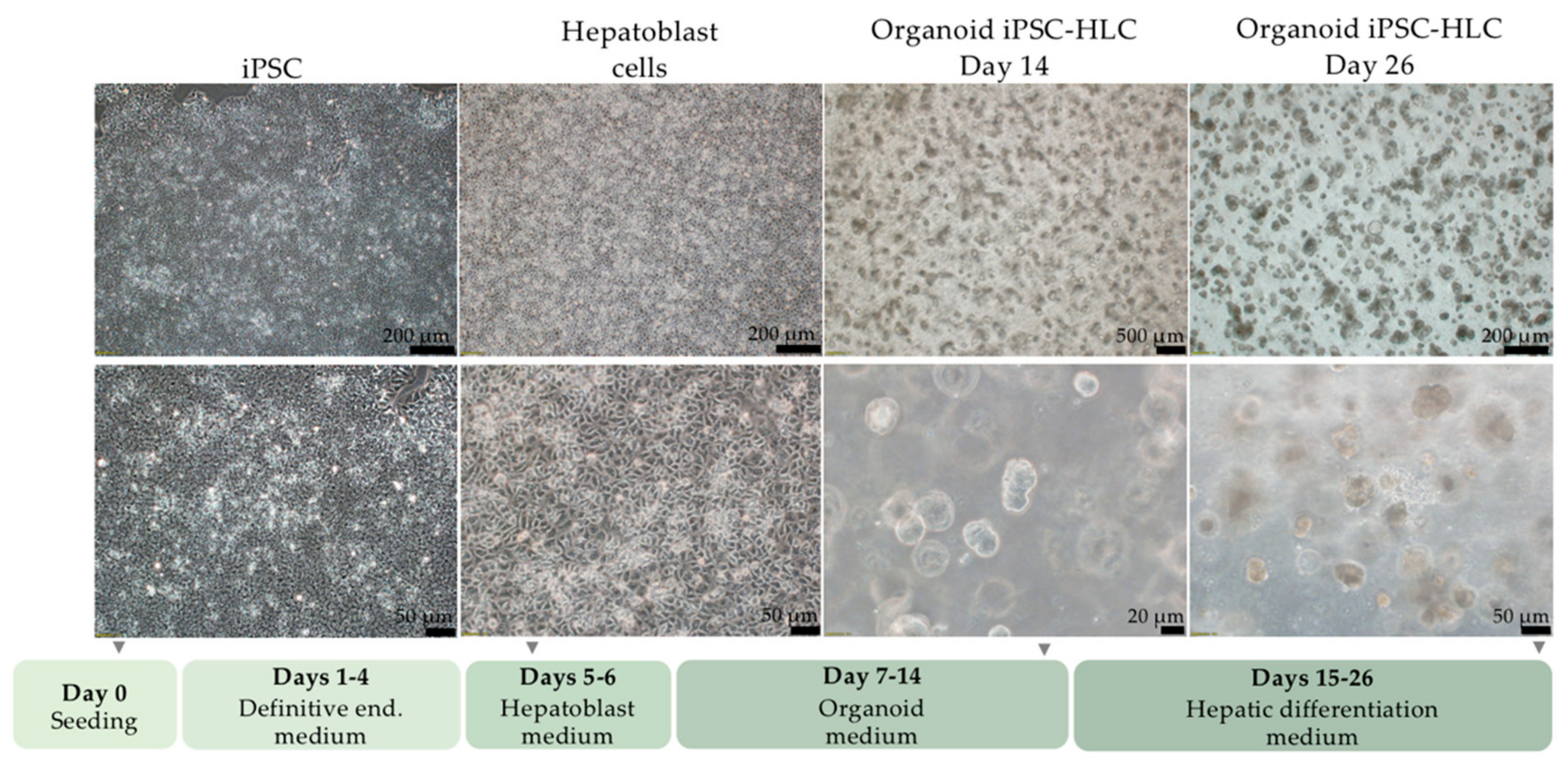
2.4. Differentiation of iPSC into Organoid iPSC-HLC
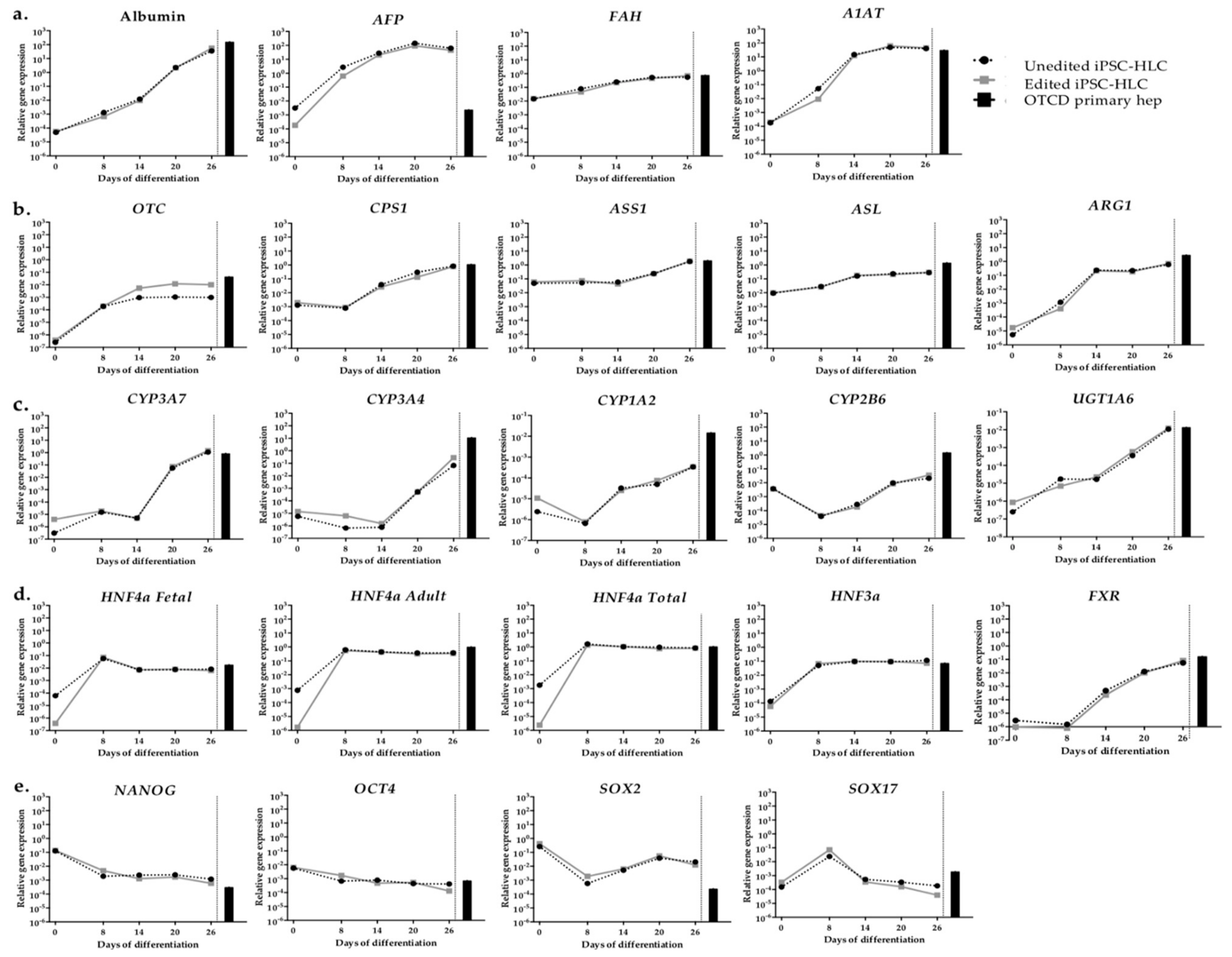
2.5. Gene Expression of Organoid iPSC-HLC
2.6. Phenotypic Characterization In Vitro
2.7. Off-Target Mutagenesis Analysis
3. Discussion
4. Materials and Methods
4.1. Cell origin and Culture
4.2. DNA and RNA Isolation
4.3. Long-Range PCR Amplification and Sequencing
4.4. Reprogramming
4.5. Gene Expression Analysis
4.6. Flow Cytometry
4.7. Alkaline Phosphatase Staining and Immunocytochemistry
4.8. PluriTest
4.9. gRNA Design
4.10. Vector Construction
4.11. Transfection
4.12. Fluorescence-Activated Cell Sorting (FACS) of Transfected Cells
4.13. Restriction Enzyme Digestion and Analysis of DNA Fragments
4.14. Single Cell Clonal Expansion and Editing Verification
4.15. OTC Transcript Amplification
4.16. Definitive Endoderm Optimization
4.17. Enzyme-Linked Immunosorbent Assay (ELISA)
4.18. In Vitro 15N-Labelled Urea Production
4.19. Off-Target Evaluation
4.20. Illustrations
4.21. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matsumoto, S.; Haberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders-update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Summar, M.L.; Koelker, S.; Freedenberg, D.; Le Mons, C.; Haberle, J.; Lee, H.S.; Kirmse, B.; European Registry; Network for Intoxication Type Metabolic Diseases; Members of the Urea Cycle Disorders Consortium. The incidence of urea cycle disorders. Mol. Genet. Metab. 2013, 110, 179–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, S.C.; Fisher, R.A.; Rubinstein, W.S.; Barranger, J.A.; Towbin, R.B.; Charron, M.; Mieles, L.; Pisarov, L.A.; Dorko, K.; Thompson, M.T.; et al. Transplantation of human hepatocytes. Transplant. Proc. 1997, 29, 2103–2106. [Google Scholar] [CrossRef]
- Stephenne, X.; Najimi, M.; Smets, F.; Reding, R.; de Ville de Goyet, J.; Sokal, E.M. Cryopreserved liver cell transplantation controls ornithine transcarbamylase deficient patient while awaiting liver transplantation. Am. J. Transplant. 2005, 5, 2058–2061. [Google Scholar] [CrossRef] [PubMed]
- Meyburg, J.; Das, A.M.; Hoerster, F.; Lindner, M.; Kriegbaum, H.; Engelmann, G.; Schmidt, J.; Ott, M.; Pettenazzo, A.; Luecke, T.; et al. One liver for four children: First clinical series of liver cell transplantation for severe neonatal urea cycle defects. Transplantation 2009, 87, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Soltys, K.A.; Setoyama, K.; Tafaleng, E.N.; Soto Gutierrez, A.; Fong, J.; Fukumitsu, K.; Nishikawa, T.; Nagaya, M.; Sada, R.; Haberman, K.; et al. Host conditioning and rejection monitoring in hepatocyte transplantation in humans. J. Hepatol. 2017, 66, 987–1000. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.M.; Wang, M.; Crane, A.M.; Brown, B.; Darlington, G.J.; Ledley, F.D. Effective cryopreservation and long-term storage of primary human hepatocytes with recovery of viability, differentiation, and replicative potential. Cell Transplant. 1995, 4, 579–586. [Google Scholar] [CrossRef]
- Mitry, R.R.; Hughes, R.D.; Dhawan, A. Progress in human hepatocytes: Isolation, culture & cryopreservation. Semin. Cell Dev. Biol. 2002, 13, 463–467. [Google Scholar] [CrossRef]
- Gramignoli, R.; Vosough, M.; Kannisto, K.; Srinivasan, R.C.; Strom, S.C. Clinical hepatocyte transplantation: Practical limits and possible solutions. Eur. Surg. Res. 2015, 54, 162–177. [Google Scholar] [CrossRef]
- Jorns, C.; Nowak, G.; Nemeth, A.; Zemack, H.; Mork, L.M.; Johansson, H.; Gramignoli, R.; Watanabe, M.; Karadagi, A.; Alheim, M.; et al. De Novo Donor-Specific HLA Antibody Formation in Two Patients With Crigler-Najjar Syndrome Type I Following Human Hepatocyte Transplantation With Partial Hepatectomy Preconditioning. Am. J. Transplant. 2016, 16, 1021–1030. [Google Scholar] [CrossRef]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Dial, S.; Shi, L.; Branham, W.; Liu, J.; Fang, J.L.; Green, B.; Deng, H.; Kaput, J.; Ning, B. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramignoli, R.; Tahan, V.; Dorko, K.; Venkataramanan, R.; Fox, I.J.; Ellis, E.C.; Vosough, M.; Strom, S.C. Rapid and sensitive assessment of human hepatocyte functions. Cell Transplant. 2014, 23, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Heslop, J.A.; Rowe, C.; Walsh, J.; Sison-Young, R.; Jenkins, R.; Kamalian, L.; Kia, R.; Hay, D.; Jones, R.P.; Malik, H.Z.; et al. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 2017, 91, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richert, L.; Liguori, M.J.; Abadie, C.; Heyd, B.; Mantion, G.; Halkic, N.; Waring, J.F. Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab. Dispos. 2006, 34, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Barahona, A.; Sanchez-Alcudia, R.; Viecelli, H.M.; Rufenacht, V.; Perez, B.; Ugarte, M.; Haberle, J.; Thony, B.; Desviat, L.R. Functional characterization of the spf/ash splicing variation in OTC deficiency of mice and man. PLoS ONE 2015, 10, e0122966. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.J.; Schuldt, B.M.; Williams, R.; Mason, D.; Altun, G.; Papapetrou, E.P.; Danner, S.; Goldmann, J.E.; Herbst, A.; Schmidt, N.O.; et al. A bioinformatic assay for pluripotency in human cells. Nat. Methods 2011, 8, U315–U354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.J.; Laurent, L.C.; Kostka, D.; Ulitsky, I.; Williams, R.; Lu, C.; Park, I.H.; Rao, M.S.; Shamir, R.; Schwartz, P.H.; et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature 2008, 455, U401–U455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasu, A.; Ikeya, M.; Yamamoto, T.; Watanabe, A.; Jin, Y.H.; Matsumoto, Y.; Hayakawa, K.; Amano, N.; Sato, S.; Osafune, K.; et al. Genetically Matched Human iPS Cells Reveal that Propensity for Cartilage and Bone Differentiation Differs with Clones, not Cell Type of Origin. PLoS ONE 2013, 8, e053771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagihara, K.; Liu, Y.; Kanie, K.; Takayama, K.; Kokunugi, M.; Hirata, M.; Fukuda, T.; Suga, M.; Nikawa, H.; Mizuguchi, H.; et al. Prediction of Differentiation Tendency Toward Hepatocytes from Gene Expression in Undifferentiated Human Pluripotent Stem Cells. Stem Cells Dev. 2016, 25, 1884–1897. [Google Scholar] [CrossRef] [Green Version]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic Memory and Preferential Lineage-Specific Differentiation in Induced Pluripotent Stem Cells Derived from Human Pancreatic Islet Beta Cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; Pampalli, S.; Lee, J.B.; McNicol, J.; Collins, T.; Draper, J.S.; Bhatia, M. Cell Fate Potential of Human Pluripotent Stem Cells Is Encoded by Histone Modifications. Cell Stem Cell 2011, 9, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Howells, K.; Phillips, A.D.; Thomas, N.S.T.; Cooper, D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009, 1. [Google Scholar] [CrossRef] [Green Version]
- Zabulica, M.; Srinivasan, R.C.; Vosough, M.; Hammarstedt, C.; Wu, T.; Gramignoli, R.; Ellis, E.; Kannisto, K.; Collin de l'Hortet, A.; Takeishi, K.; et al. Guide to the Assessment of Mature Liver Gene Expression in Stem Cell-Derived Hepatocytes. Stem Cells Dev. 2019, 28, 907–919. [Google Scholar] [CrossRef] [Green Version]
- Marongiu, F.; Gramignoli, R.; Dorko, K.; Miki, T.; Ranade, A.R.; Serra, M.P.; Doratiotto, S.; Sini, M.; Sharma, S.; Mitamura, K.; et al. Hepatic Differentiation of Amniotic Epithelial Cells. Hepatology 2011, 53, 1719–1729. [Google Scholar] [CrossRef] [Green Version]
- Takebe, T.; Zhang, R.R.; Koike, H.; Kimura, M.; Yoshizawa, E.; Enomura, M.; Koike, N.; Sekine, K.; Taniguchi, H. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat. Protoc. 2014, 9, 396–409. [Google Scholar] [CrossRef]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.K.; et al. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 2013, 34, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Gieseck, R.L., 3rd; Hannan, N.R.; Bort, R.; Hanley, N.A.; Drake, R.A.; Cameron, G.W.; Wynn, T.A.; Vallier, L. Maturation of induced pluripotent stem cell derived hepatocytes by 3D-culture. PLoS ONE 2014, 9, e86372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Haberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Li, S.B.; Guo, J.Y.; Ying, Z.F.; Chen, S.; Yang, L.; Chen, K.S.; Long, Q.; Qin, D.J.; Pei, D.Q.; Liu, X.G. Valproic Acid-Induced Hepatotoxicity in Alpers Syndrome Is Associated With Mitochondrial Permeability Transition Pore Opening-Dependent Apoptotic Sensitivity in an Induced Pluripotent Stem Cell Model. Hepatology 2015, 61, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Yoshitoshi-Uebayashi, E.Y.; Toyoda, T.; Yasuda, K.; Kotaka, M.; Nomoto, K.; Okita, K.; Yasuchika, K.; Okamoto, S.; Takubo, N.; Nishikubo, T.; et al. Modelling urea-cycle disorder citrullinemia type 1 with disease-specific iPSCs. Biochem. Biophys. Res. Commun. 2017, 486, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Tapia, N.; Scholer, H.R. Molecular Obstacles to Clinical Translation of iPSCs. Cell Stem Cell 2016, 19, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Raju, R.; Chau, D.; Notelaers, T.; Myers, C.L.; Verfaillie, C.M.; Hu, W.S. In Vitro Pluripotent Stem Cell Differentiation to Hepatocyte Ceases Further Maturation at an Equivalent Stage of E15 in Mouse Embryonic Liver Development. Stem Cells Dev. 2018, 27, 910–921. [Google Scholar] [CrossRef]
- Lee, P.C.; Truong, B.; Vega-Crespo, A.; Gilmore, W.B.; Hermann, K.; Angarita, S.A.; Tang, J.K.; Chang, K.M.; Wininger, A.E.; Lam, A.K.; et al. Restoring Ureagenesis in Hepatocytes by CRISPR/Cas9-mediated Genomic Addition to Arginase-deficient Induced Pluripotent Stem Cells. Mol. Ther. Nucleic Acids 2016, 5, e394. [Google Scholar] [CrossRef]
- Nakamura, T.; Morishige, S.; Ozawa, H.; Kuboyama, K.; Yamasaki, Y.; Oya, S.; Yamaguchi, M.; Aoyama, K.; Seki, R.; Mouri, F.; et al. Successful correction of factor V deficiency of patient-derived iPSCs by CRISPR/Cas9-mediated gene editing. Haemophilia 2020, 26, 826–833. [Google Scholar] [CrossRef]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets (vol 14, pg 607, 2017). Nat. Methods 2018, 15, 394. [Google Scholar] [CrossRef] [Green Version]
- Wienert, B.; Wyman, S.K.; Richardson, C.D.; Yeh, C.D.; Akcakaya, P.; Porritt, M.J.; Morlock, M.; Vu, J.T.; Kazane, K.R.; Watry, H.L.; et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 2019, 364, 286. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deinsberger, J.; Reisinger, D.; Weber, B. Global trends in clinical trials involving pluripotent stem cells: A systematic multi-database analysis. NPJ Regen. Med. 2020, 5. [Google Scholar] [CrossRef]
- Gramignoli, R.; Green, M.L.; Tahan, V.; Dorko, K.; Skvorak, K.J.; Marongiu, F.; Zao, W.C.; Venkataramanan, R.; Ellis, E.C.S.; Geller, D.; et al. Development and Application of Purified Tissue Dissociation Enzyme Mixtures for Human Hepatocyte Isolation. Cell Transplant. 2012, 21, 1245–1260. [Google Scholar] [CrossRef]
- Vosough, M.; Ravaioli, F.; Zabulica, M.; Capri, M.; Garagnani, P.; Franceschi, C.; Piccand, J.; Kraus, M.R.; Kannisto, K.; Gramignoli, R.; et al. Applying hydrodynamic pressure to efficiently generate induced pluripotent stem cells via reprogramming of centenarian skin fibroblasts. PLoS ONE 2019, 14, e0215490. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Allegri, G.; Deplazes, S.; Grisch-Chan, H.M.; Mathis, D.; Fingerhut, R.; Haberle, J.; Thony, B. A simple dried blood spot-method for in vivo measurement of ureagenesis by gas chromatography-mass spectrometry using stable isotopes. Clin. Chim. Acta 2017, 464, 236–243. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zabulica, M.; Jakobsson, T.; Ravaioli, F.; Vosough, M.; Gramignoli, R.; Ellis, E.; Rooyackers, O.; Strom, S.C. Gene Editing Correction of a Urea Cycle Defect in Organoid Stem Cell Derived Hepatocyte-like Cells. Int. J. Mol. Sci. 2021, 22, 1217. https://doi.org/10.3390/ijms22031217
Zabulica M, Jakobsson T, Ravaioli F, Vosough M, Gramignoli R, Ellis E, Rooyackers O, Strom SC. Gene Editing Correction of a Urea Cycle Defect in Organoid Stem Cell Derived Hepatocyte-like Cells. International Journal of Molecular Sciences. 2021; 22(3):1217. https://doi.org/10.3390/ijms22031217
Chicago/Turabian StyleZabulica, Mihaela, Tomas Jakobsson, Francesco Ravaioli, Massoud Vosough, Roberto Gramignoli, Ewa Ellis, Olav Rooyackers, and Stephen C. Strom. 2021. "Gene Editing Correction of a Urea Cycle Defect in Organoid Stem Cell Derived Hepatocyte-like Cells" International Journal of Molecular Sciences 22, no. 3: 1217. https://doi.org/10.3390/ijms22031217
APA StyleZabulica, M., Jakobsson, T., Ravaioli, F., Vosough, M., Gramignoli, R., Ellis, E., Rooyackers, O., & Strom, S. C. (2021). Gene Editing Correction of a Urea Cycle Defect in Organoid Stem Cell Derived Hepatocyte-like Cells. International Journal of Molecular Sciences, 22(3), 1217. https://doi.org/10.3390/ijms22031217