
CD1d-Dependent iNKT Cells Control DSS-Induced Colitis in a Mouse Model of IFNγ-Mediated Hyperinflammation by Increasing IL22-Secreting ILC3 Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
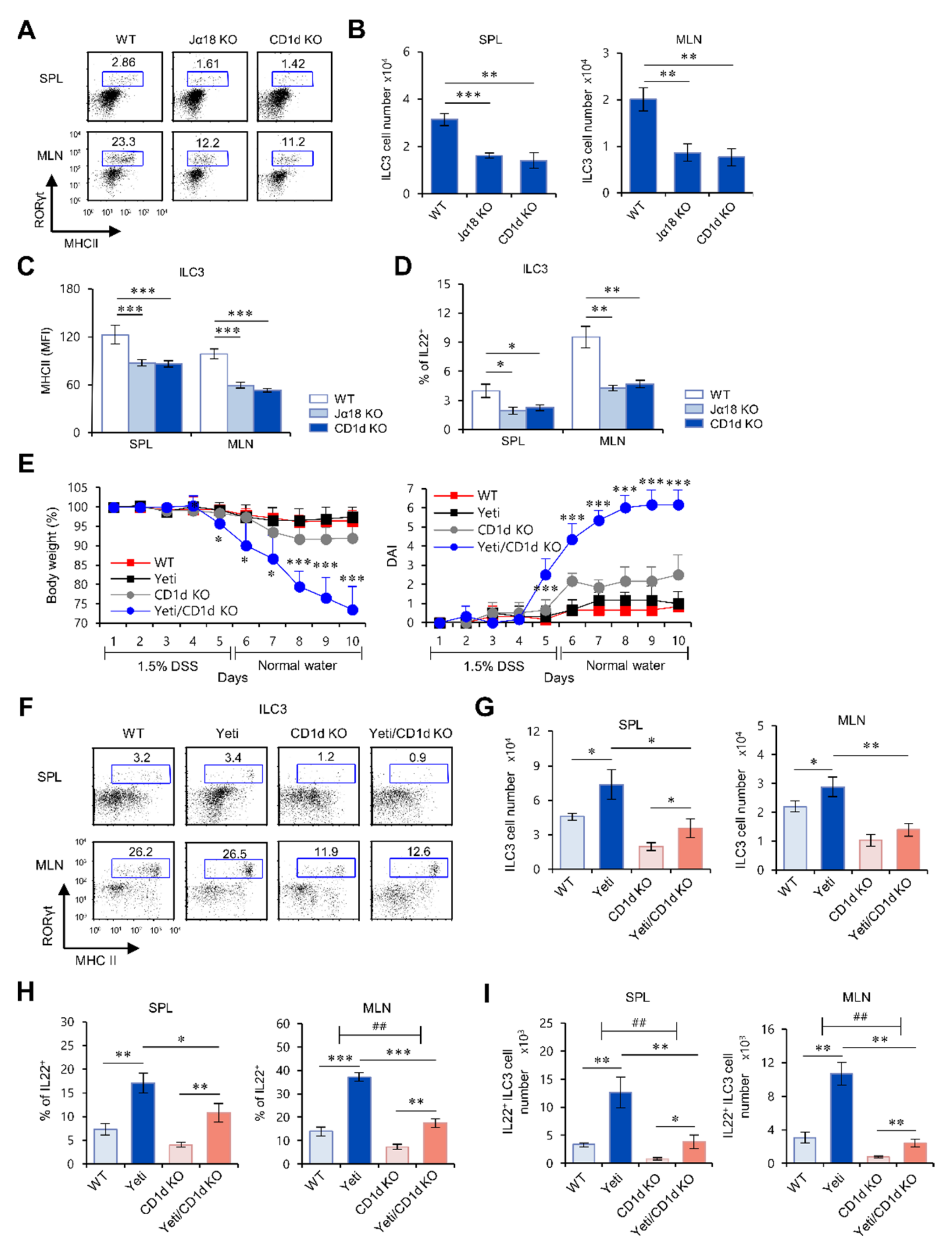
2.1. Yeti Mice Display a Dramatic Increase of IL22-Producing ILC3s during DSS-Induced Intestinal Inflammation
2.2. The Frequency and Function of ILC3s in the MLN of Yeti Mice Are Dependent on CD1d-Dependent iNKT Cells
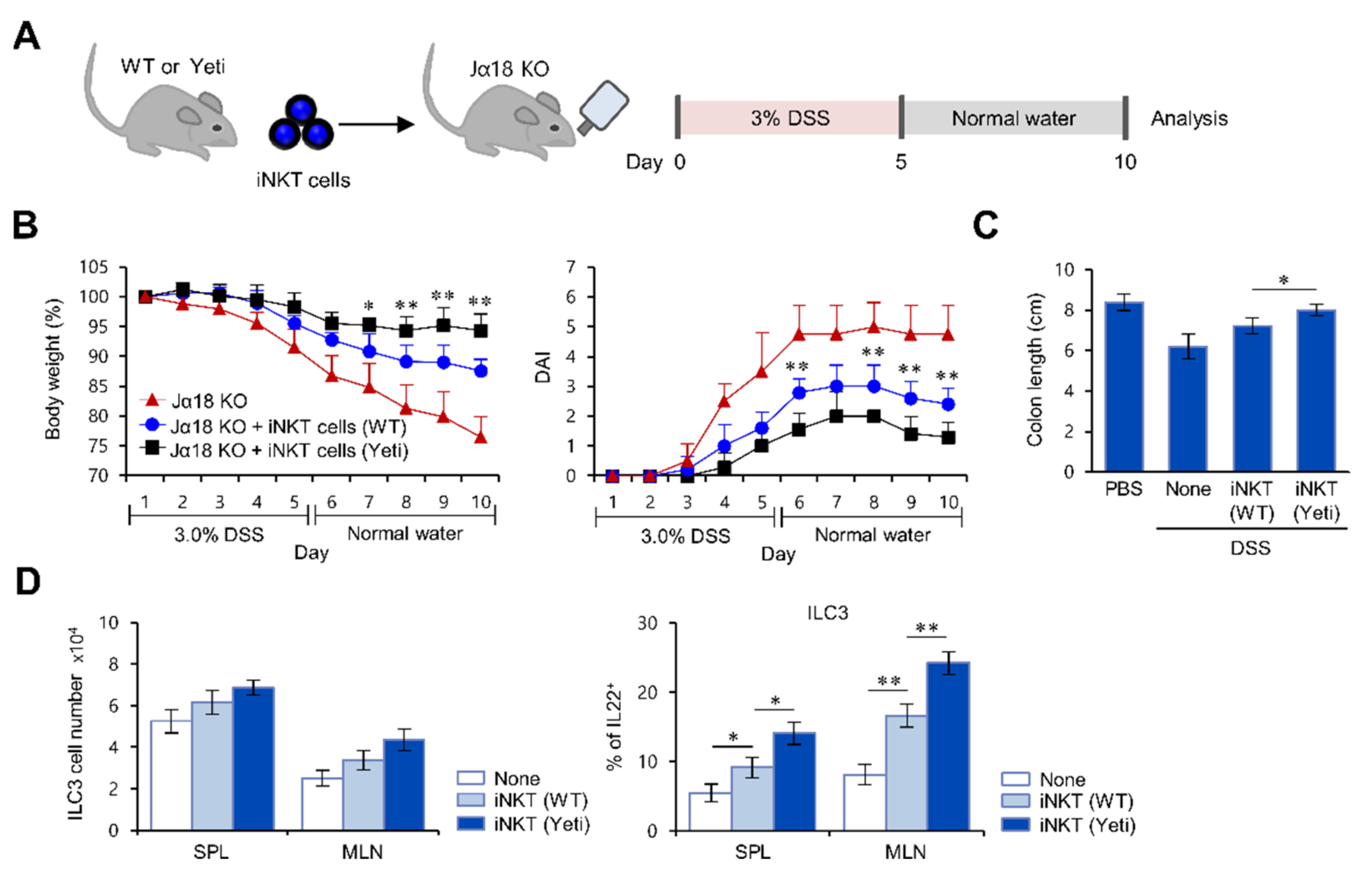
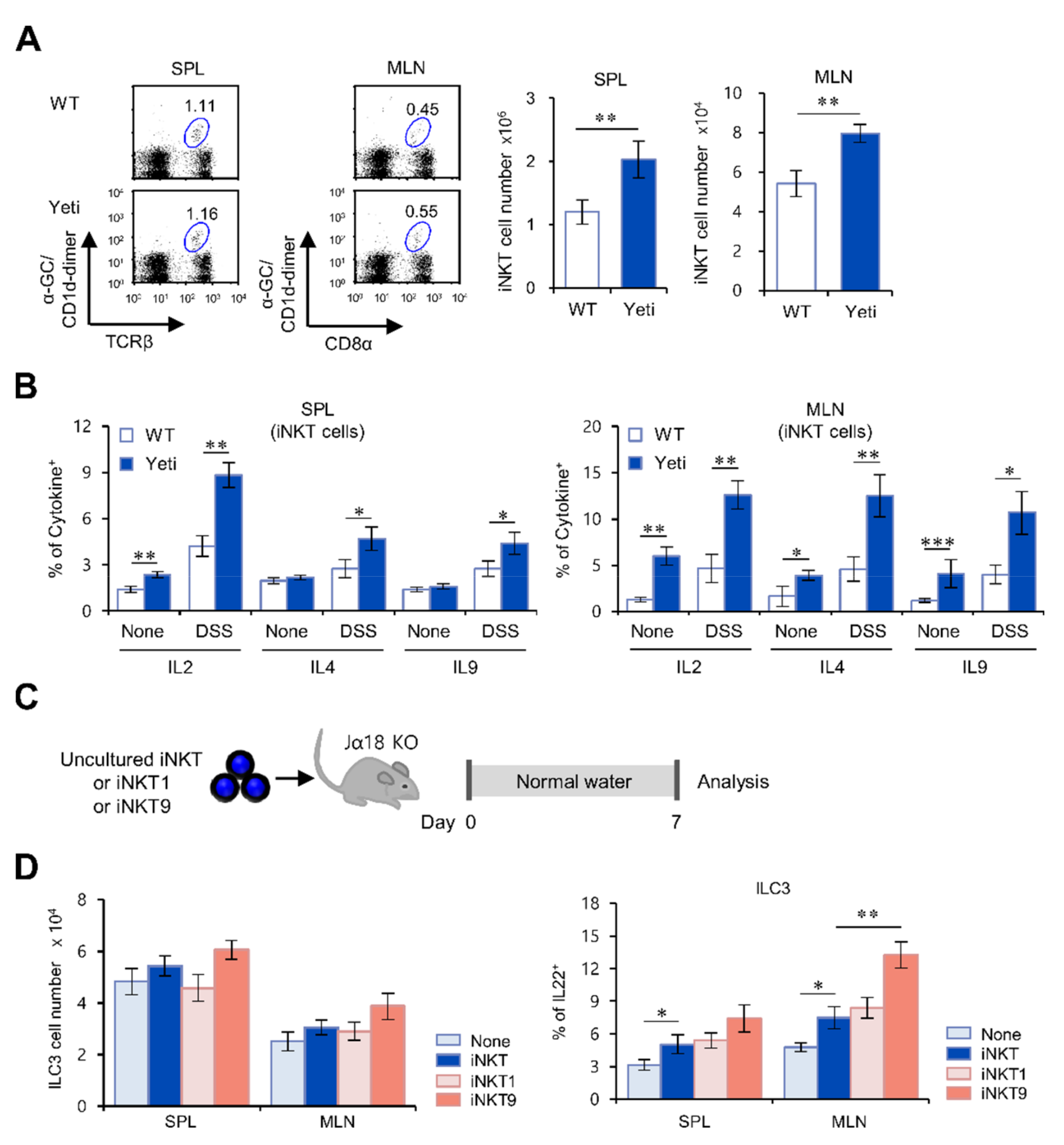
2.3. Suppressive Effects of iNKT Cells on IFNγ-Mediated Intestinal Inflammation Are Closely Associated with Increased IL22 Production by ILC3s
2.4. The IL9-Producing Subset of iNKT Cells Up-Regulates IL22-Producing ILC3s in IFNγ-Mediated Intestinal Inflammation
3. Discussion
4. Materials and Methods
4.1. Mice and Reagents
4.2. Induction of Colonic Inflammation
4.3. Cell Culture and Cell Enrichment by Magnetically Activated Cell Sorting(MACS)
4.4. CD1d/α-GalCer Dimer Staining for iNKT Cells
4.5. In Vitro iNKT Cell Differentiation
4.6. Flow Cytometry
4.7. Intracellular Cytokine Staining
4.8. Isolation of Colon MLN Leukocytes
4.9. Histology
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nat. Cell Biol. 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, R.L.; Liang, H.E.; Bao, K.; Price, A.E.; Mohrs, M.; Kelly, B.L.; Locksley, R.M. A novel model for IFN-gamma-mediated autoinflammatory syndromes. J. Immunol. 2015, 194, 2358–2368. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Park, H.J.; Cheon, J.H.; Wu, L.; Van Kaer, L.; Hong, S. iNKT Cells Suppress Pathogenic NK1.1+CD8+ T Cells in DSS-Induced Colitis. Front. Immunol. 2018, 9, 2168. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Son, S.; Oliveira, S.C.; Barber, G.N. STING-Dependent Signaling Underlies IL-10 Controlled Inflammatory Colitis. Cell Rep. 2017, 21, 3873–3884. [Google Scholar] [CrossRef] [Green Version]
- Van Kaer, L.; Wu, L. Therapeutic Potential of Invariant Natural Killer T Cells in Autoimmunity. Front. Immunol. 2018, 9, 519. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Driver, J.P.; Van Kaer, L. The Role of Autophagy in iNKT Cell Development. Front. Immunol. 2018, 9, 2653. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.W.; Im, W.; Kim, M.; Van Kaer, L.; Hong, S. iNKT Cell Activation Exacerbates the Development of Huntington’s Disease in R6/2 Transgenic Mice. Mediators Inflamm. 2019, 2019, 3540974. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, H.J.; van Kaer, L.; Hong, S.; Hong, S. Graphene oxide polarizes iNKT cells for production of TGFbeta and attenuates inflammation in an iNKT cell-mediated sepsis model. Sci. Rep. 2018, 8, 10081. [Google Scholar]
- Park, H.J.; Lee, S.W.; Park, S.H.; Van Kaer, L.; Hong, S. Selective Expansion of Double Negative iNKT Cells Inhibits the De-velopment of Atopic Dermatitis in Vα14 TCR Transgenic NC/Nga Mice by Increasing Memory-Type CD8+ T and Reg-ulatory CD4+ T Cells. J. Invest. Dermatol. 2020. [Google Scholar] [CrossRef]
- Kim, H.S.; Chung, D.H. IL-9-producing invariant NKT cells protect against DSS-induced colitis in an IL-4-dependent manner. Mucosal Immunol. 2013, 6, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, S.; Lochner, M.; Satoh-Takayama, N.; Dulauroy, S.; Bérard, M.; Kleinschek, M.; Cua, D.; Di Santo, J.P.; Eberl, G. ROR-γt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011, 12, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackley, E.C.; Houston, S.; Marriott, C.L.; Halford, E.E.; Lucas, B.; Cerovic, V.; Filbey, K.J.; Maizels, R.M.; Hepworth, M.R.; Sonnenberg, G.F.; et al. CCR7-dependent trafficking of RORγ+ ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat. Commun. 2015, 6, 5862. [Google Scholar] [CrossRef] [PubMed]
- De Guinoa, J.S.; Jimeno, R.; Farhadi, N.; Jervis, P.J.; Cox, L.R.; Besra, G.S.; Barral, P. CD1d-mediated activation of group 3 innate lymphoid cells drives IL-22 production. EMBO Rep. 2017. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, H.J.; Pei, Y.; Yeo, Y.; Hong, S. Topical application of zwitterionic chitosan suppresses neutrophil-mediated acute skin inflammation. Int. J. Biol. Macromol. 2020, 158, 1184–1193. [Google Scholar] [CrossRef]
- Ju, A.; Lee, S.W.; Lee, Y.E.; Han, K.C.; Kim, J.C.; Shin, S.C.; Park, H.J.; EunKyeong Kim, E.; Hong, S.; Jang, M. A carrier-free mul-tiplexed gene editing system applicable for suspension cells. Biomaterials 2019, 217, 119298. [Google Scholar] [CrossRef]
- Abidi, A.; Laurent, T.; Bériou, G.; Bouchet-Delbos, L.; Fourgeux, C.; Louvet, C.; Triki-Marrakchi, R.; Poschmann, J.; Josien, R.; Martin, J.C. Characterization of Rat ILCs Reveals ILC2 as the Dominant Intestinal Subset. Front. Immunol. 2020, 11, 255. [Google Scholar] [CrossRef] [Green Version]
- Diehl, G.E.; Longman, R.S.; Zhang, J.X.; Breart, B.; Galan, C.; Cuesta, A.; Schwab, S.R.; Littman, D.R. Microbiota restricts traf-ficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature 2013, 494, 116–120. [Google Scholar] [CrossRef]
- Mosconi, I.; Dubey, L.K.; Volpe, B.; Esser-von Bieren, J.; Zaiss, M.M.; Lebon, L.; Massacand, J.C.; Harris, N.L. Parasite Proximity Drives the Expansion of Regulatory T Cells in Peyer’s Patches following Intestinal Helminth Infection. Infect. Immun. 2015, 3, 3657–3665. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Wang, H.; Starrett, G.J.; Phuong, V.; Jameson, S.C.; Hogquist, K.A. Tissue-Specific Distribution of iNKT Cells Impacts Their Cytokine Response. Immunity 2015, 43, 566–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Qiu, J.; Tu, T.; Yang, X.; Deng, L.; Anders, R.A.; Zhou, L.; Fu, Y.-X. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 2014, 40, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Li, Y.; Zhang, J.; Chang, R.; Li, J.; Huo, L. IL-9 promotes the pathogenesis of ulcerative colitis through STAT3/SOCS3 signaling. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.H.; Zheng, Z.N.; Xiao, C.X.; Liu, X.; Lin, X.Q. IL-22 relieves sepsis-induced liver injury via activating JAK/STAT3 signaling pathway. J. Biol. Regul. Homeost. Agents 2020, 34, 1719–1727. [Google Scholar]
- Della Valle, L.; Gatta, A.; Farinelli, A.; Scarano, G.; Lumaca, A.; Tinari, N.; Cipollone, F.; Paganelli, R.; Di Gioacchino, M.F. Aller-gooncology: An expanding research area. J. Biol. Regul. Homeost. Agents 2020, 34, 319–326. [Google Scholar]
- Gorski, S.A.; Hahn, Y.S.; Braciale, T.J. Braciale, Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during in-fluenza virus infection. PLoS Pathog. 2013, 9, e1003615. [Google Scholar] [CrossRef]
- Trittel, S.; Vashist, N.; Ebensen, T.; Chambers, B.J.; Guzmán, C.A.; Riese, P. Invariant NKT Cell-Mediated Modulation of ILC1s as a Tool for Mucosal Immune Intervention. Front. Immunol. 2019, 10, 1849. [Google Scholar] [CrossRef] [Green Version]
- Groh, V.; Steinle, A.; Bauer, S.; Spies, T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 1998, 279, 1737–1740. [Google Scholar] [CrossRef]
- Vadstrup, K.; Galsgaard, E.D.; Jensen, H.; Lanier, L.L.; Ryan, J.C.; Chen, S.Y.; Nolan, G.P.; Vester-Andersena, M.K.; Pedersen, J.S.; Gerwien, J.; et al. NKG2D ligand expression in Crohn’s disease and NKG2D-dependent stimulation of CD8+ T cell migration. Exp. Mol. Pathol. 2017, 103, 56–70. [Google Scholar] [CrossRef] [Green Version]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.J.; Lee, S.W.; Van Kaer, L.; Hong, S. CD1d-Dependent iNKT Cells Control DSS-Induced Colitis in a Mouse Model of IFNγ-Mediated Hyperinflammation by Increasing IL22-Secreting ILC3 Cells. Int. J. Mol. Sci. 2021, 22, 1250. https://doi.org/10.3390/ijms22031250
Park HJ, Lee SW, Van Kaer L, Hong S. CD1d-Dependent iNKT Cells Control DSS-Induced Colitis in a Mouse Model of IFNγ-Mediated Hyperinflammation by Increasing IL22-Secreting ILC3 Cells. International Journal of Molecular Sciences. 2021; 22(3):1250. https://doi.org/10.3390/ijms22031250
Chicago/Turabian StylePark, Hyun Jung, Sung Won Lee, Luc Van Kaer, and Seokmann Hong. 2021. "CD1d-Dependent iNKT Cells Control DSS-Induced Colitis in a Mouse Model of IFNγ-Mediated Hyperinflammation by Increasing IL22-Secreting ILC3 Cells" International Journal of Molecular Sciences 22, no. 3: 1250. https://doi.org/10.3390/ijms22031250