
Hyaluronic Acid-Targeted Stimuli-Sensitive Nanomicelles Co-Encapsulating Paclitaxel and Ritonavir to Overcome Multi-Drug Resistance in Metastatic Breast Cancer and Triple-Negative Breast Cancer Cells



Abstract
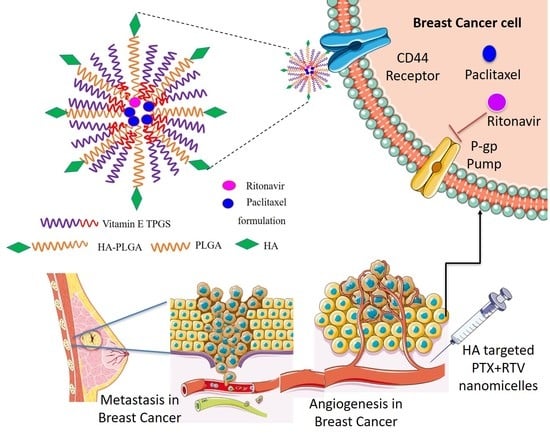
:
1. Introduction
2. Results and Discussion
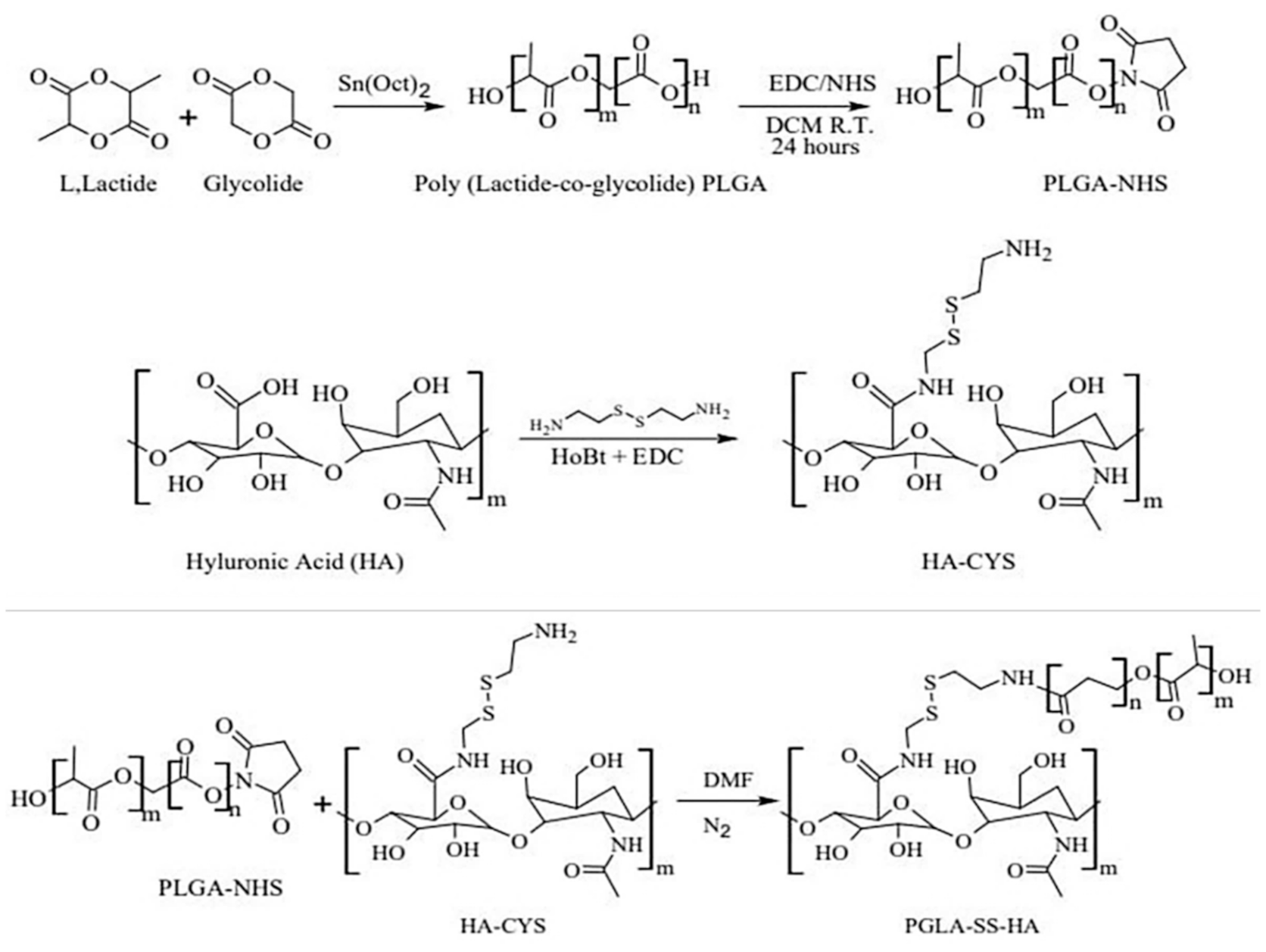
2.1. Synthesis and Characterization of HA-ss-PLGA Copolymer
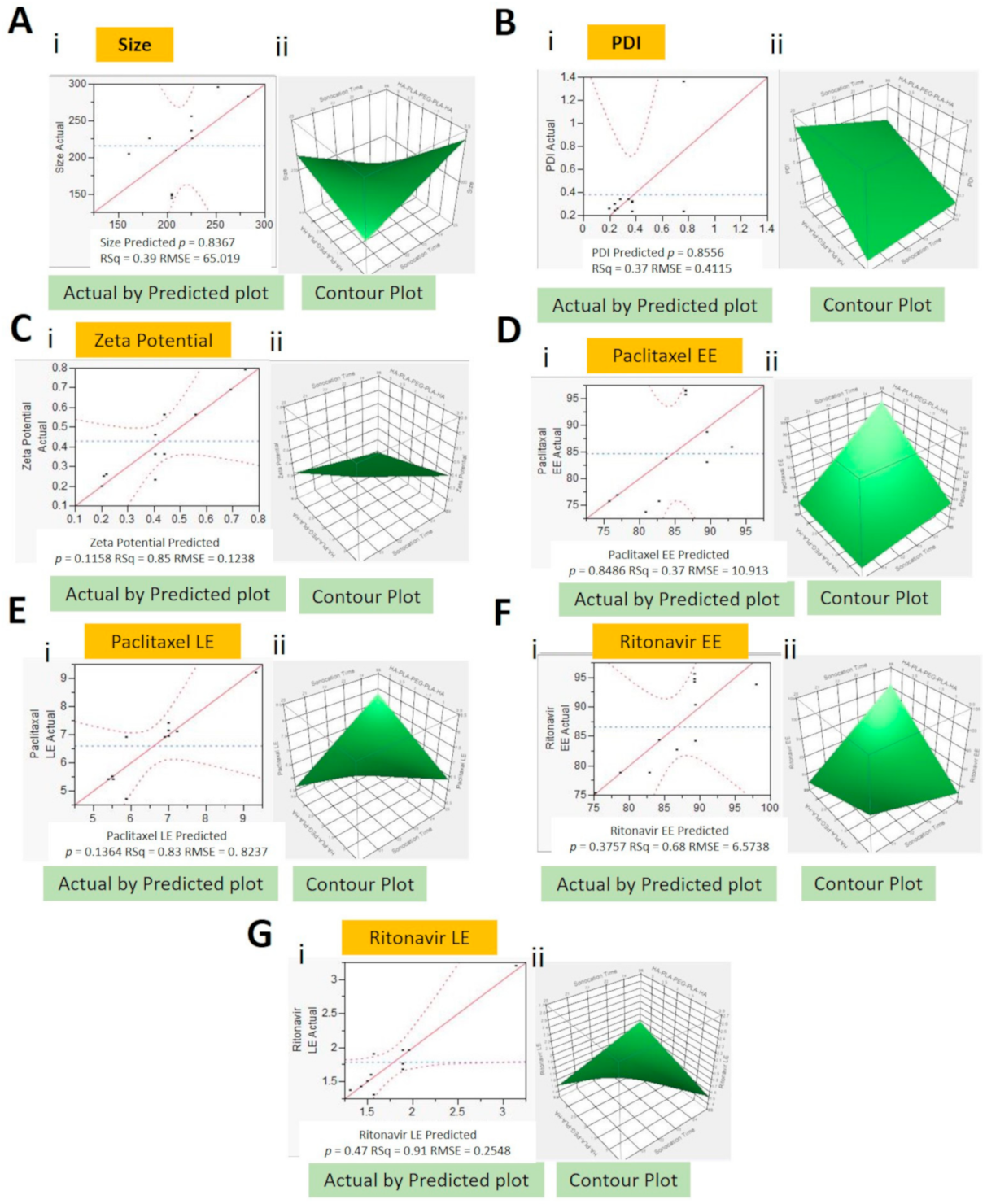
2.2. Design of Experiment and Formulation Optimization
- Y1 = 1005.4 − 53.6 × X1 + 266.4 × X2 + 185.8 × X3 × X1 − (X2 × 11.1) × X1 − (X3 × 7.9) × X2 − (X3 × 8.3)
- Y2 = 0.23 + 0.02 × x1 + 0.06 × X2 + 0.05 × X3 × X1 + (X2 × 0.01) × X1 + (X3 × 0.002) × X2 + (X3 × 0.06)
- Y3 = 0.2 +0.04 × X1 − 0.6 × X2 + 0.5 × X3 × X1 − (X2 × 0.3) × X2 + (X3 × 0.05)
- Y4 = 184 − 5.2 × X1 − 22.1 × X2 − 26.2 × X3 × X1 + (X2 × 0.74) × X1 + (X3 × 1.34) × X2 + (X3 × 1.83)
- Y5 = 57.3 − 2.2 × X1 − 801 × X2 − 9.8 × X3 × X1 + (X2 × 0.4) × X2 + (X3 × 0.5)
- Y6 = 355 − 11.3 × X1 − 57.2 × X2 − 52.8 × X3 × X1 + (X2 × 1.8) × X1 + (X! × 2.2) × X2 + (X3 × 4.6)
- Y7 = 20.3 − 0.74 × X1 − 3.23 × X2 − 3.11 × X3 × X1 + (X3 × 0.12) × X2 + (X3 × 0.21)
- where Y1—size (nm), Y2—PDI, Y3—zeta potential (mV), Y4—PTX EE (%), Y5—PTX LE (%), Y6—RTV EE (%), Y7—RTV LE (%), X1—sonication time (min), X2—HA-PLGA (wt%), and X3—Vit E-TPGS (wt%)
2.3. Formulation Characterization
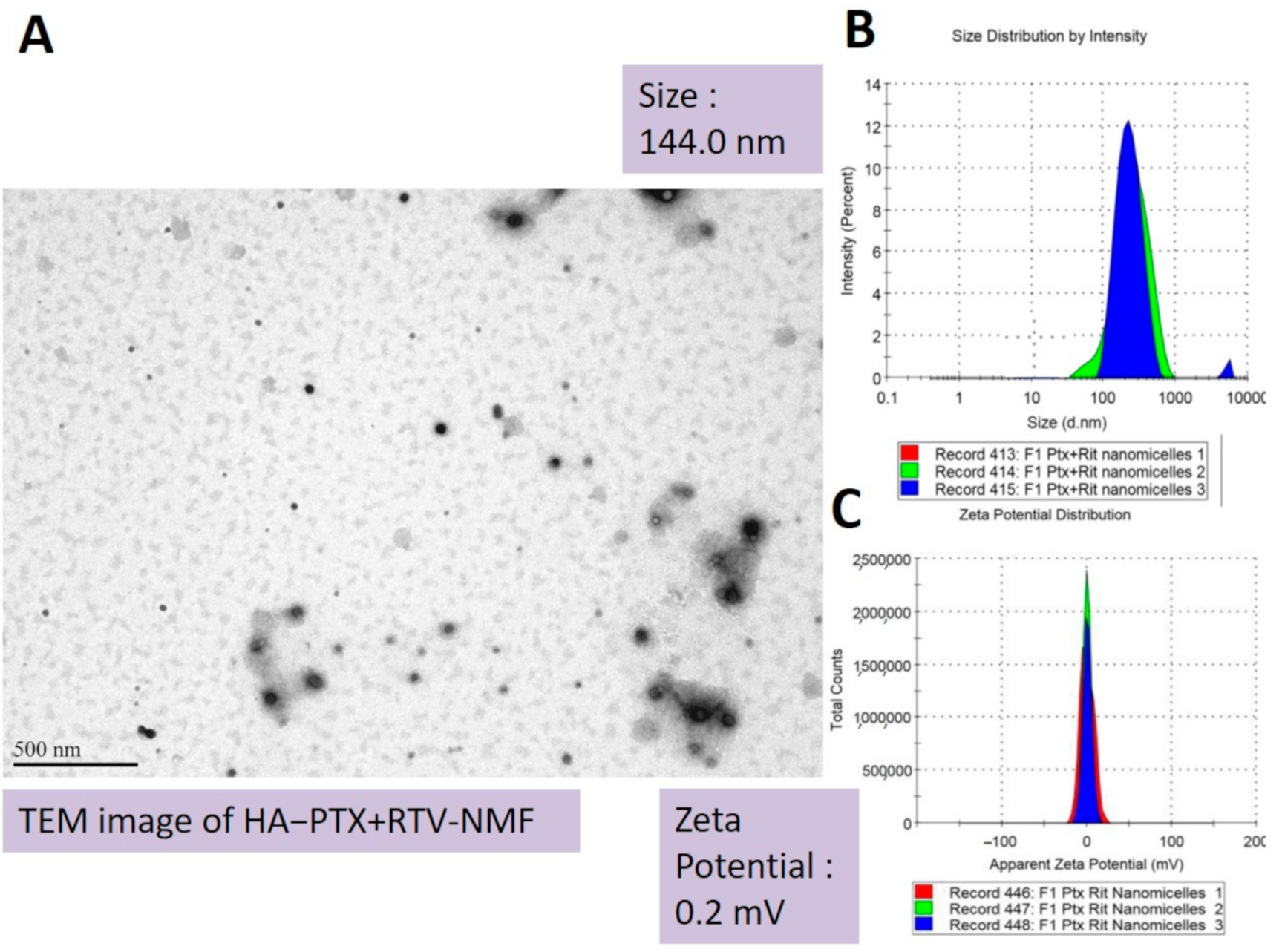
2.3.1. Size, PDI, and Zeta Potential
2.3.2. Entrapment and Loading Efficiencies
2.4. Critical Micellar Concentration
2.5. Effect of Dilution and Temperature on Storage Stability
2.6. Temperature Stability Studies
2.7. Dissolution and Drug Release
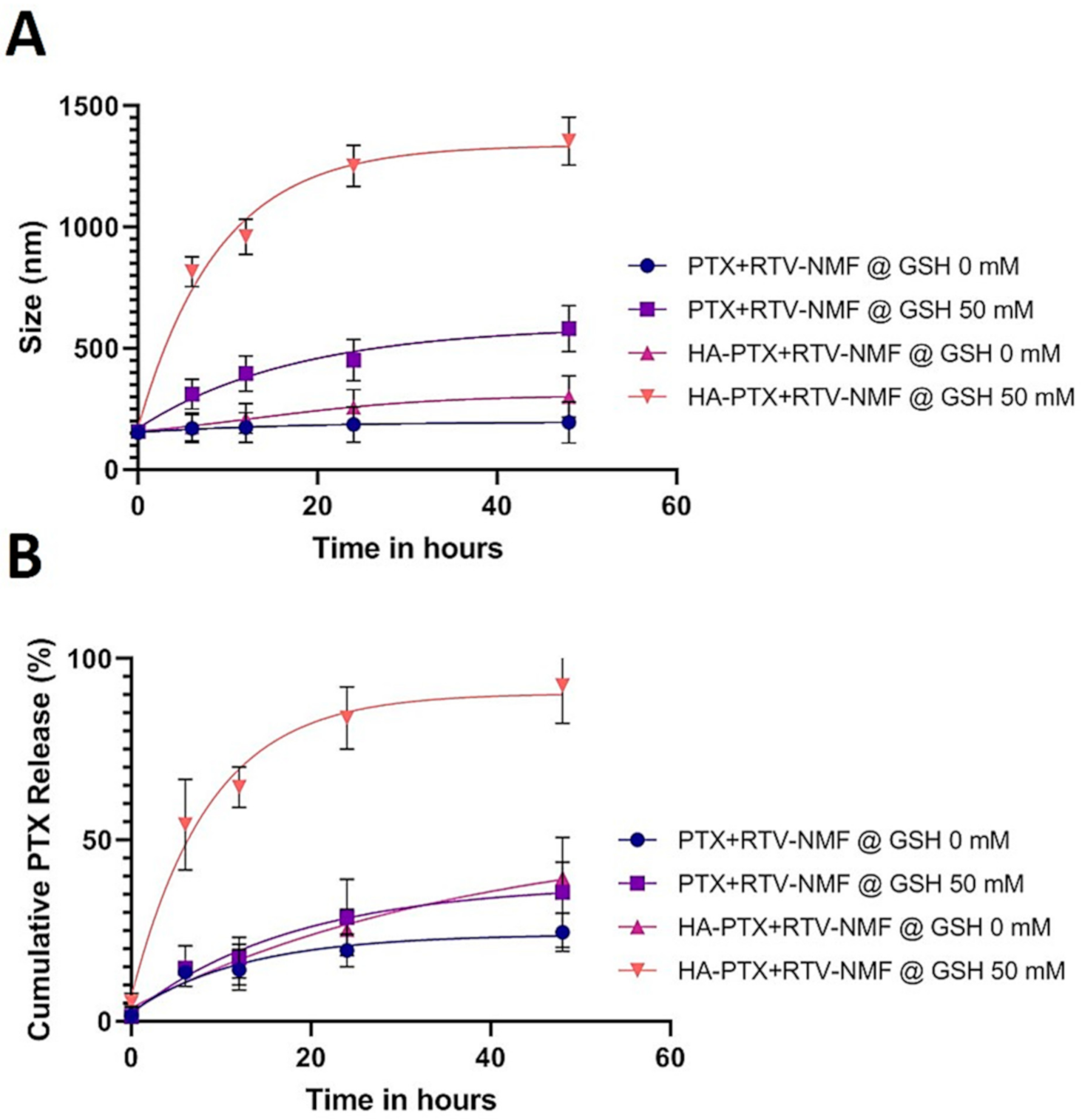
2.8. Nanomicelles in Reduction Stimulated Environment
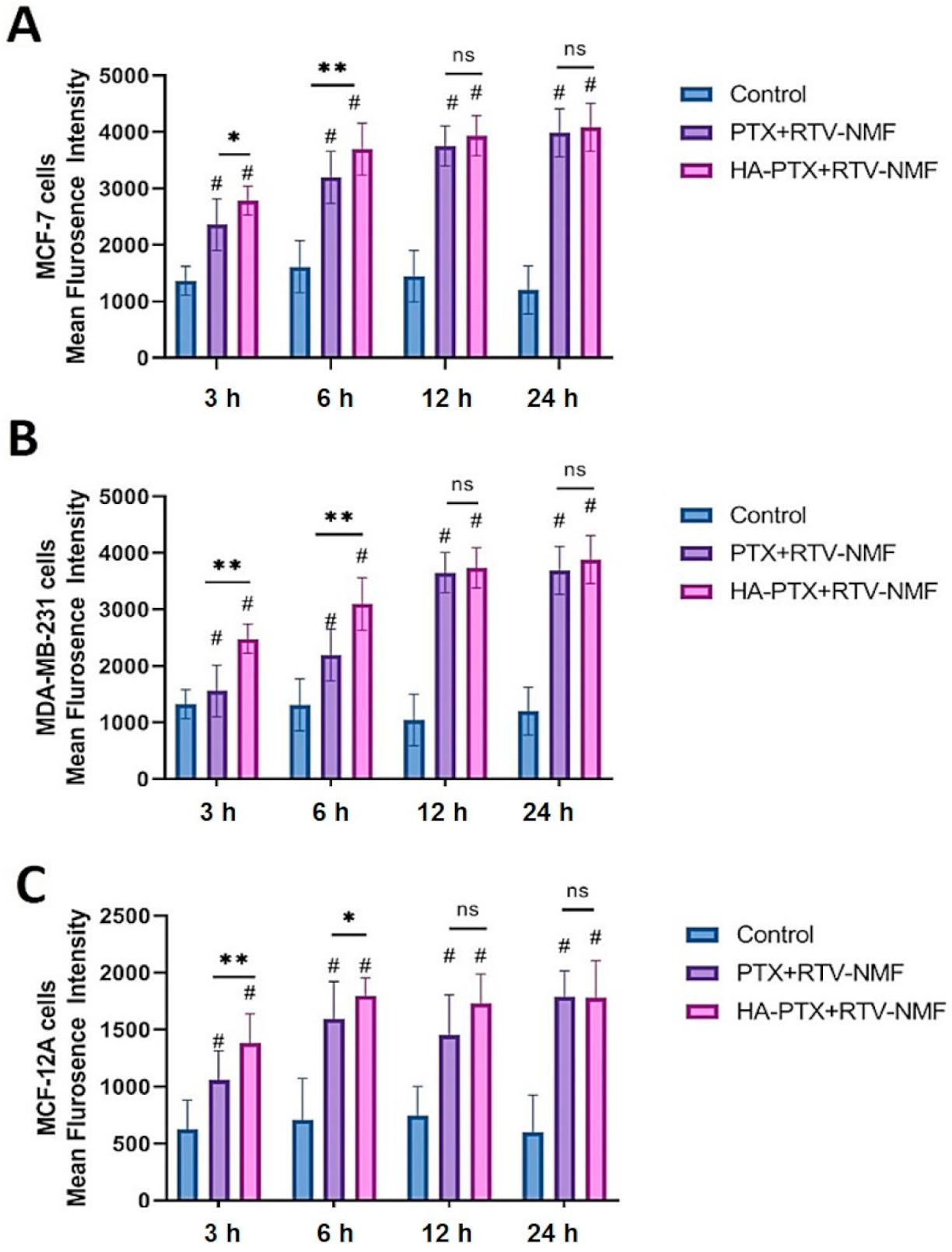
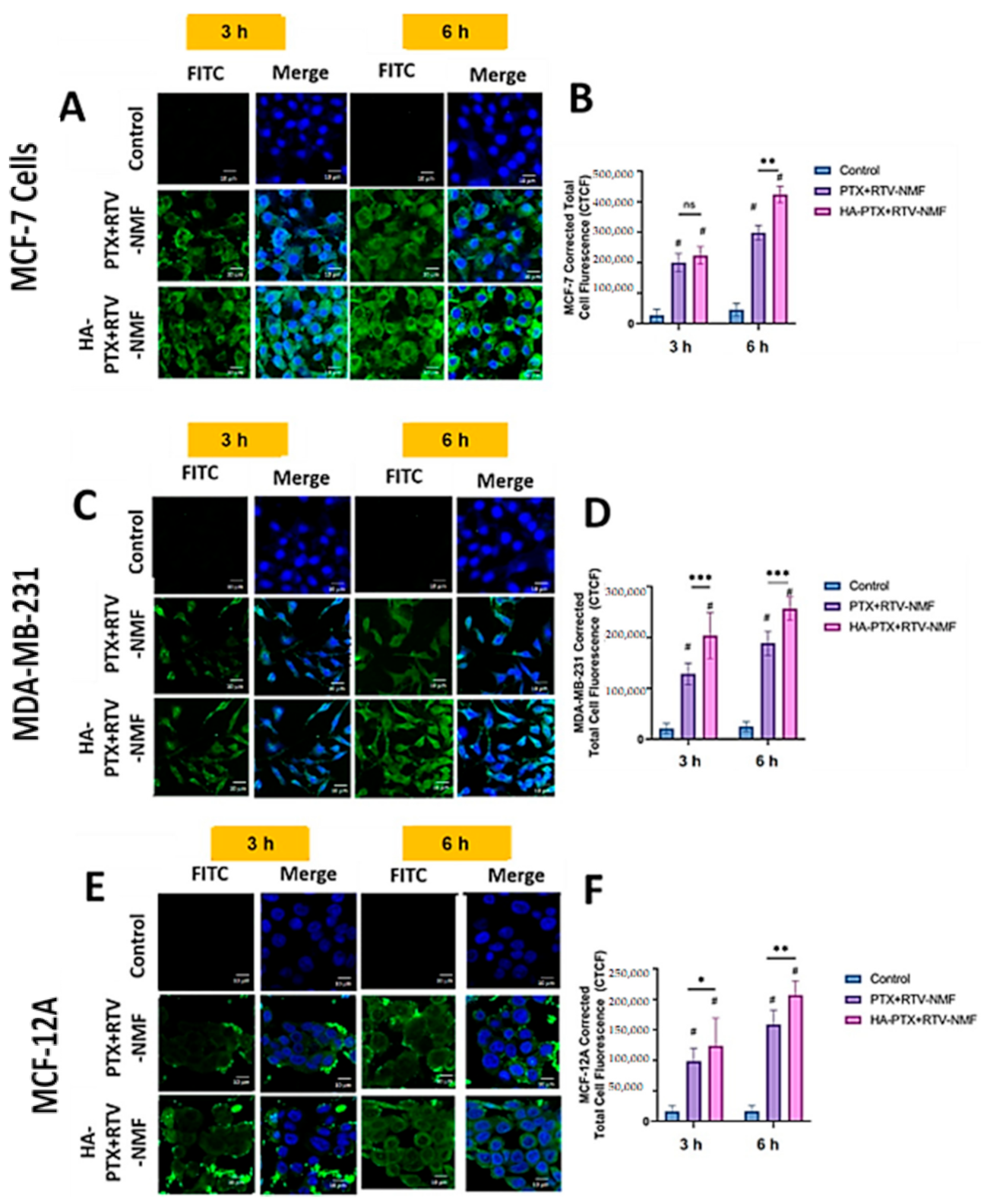
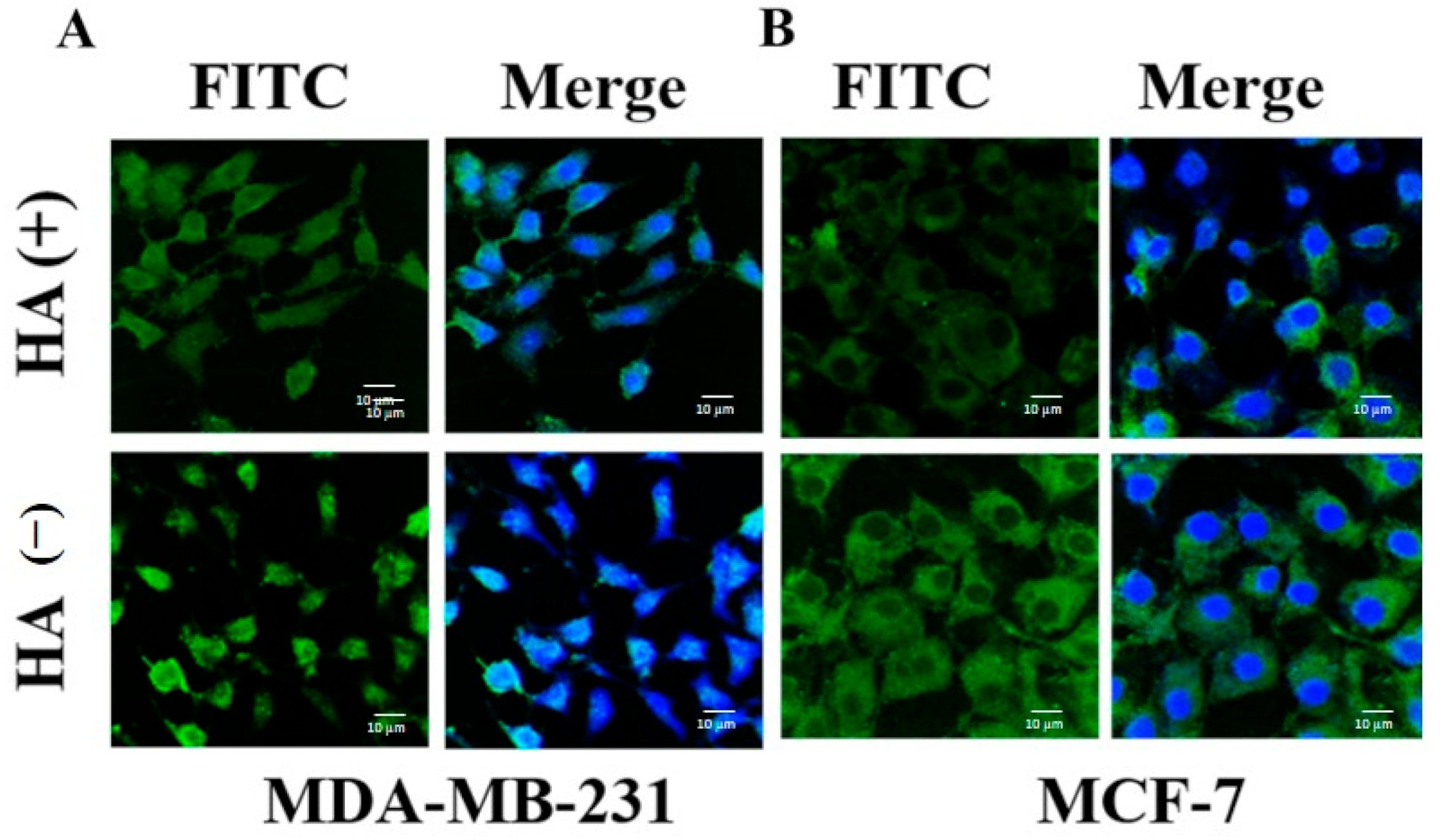
2.9. Cellular Uptake and Intracellular Distribution Study
2.9.1. Cellular Uptake by fluorescence-assisted cell sorting (FACS)
2.9.2. Intracellular Distribution Using Confocal Microscopy
2.10. In Vitro Cytotoxicity Determination
2.11. In Vitro Determination of Targeting Efficiency
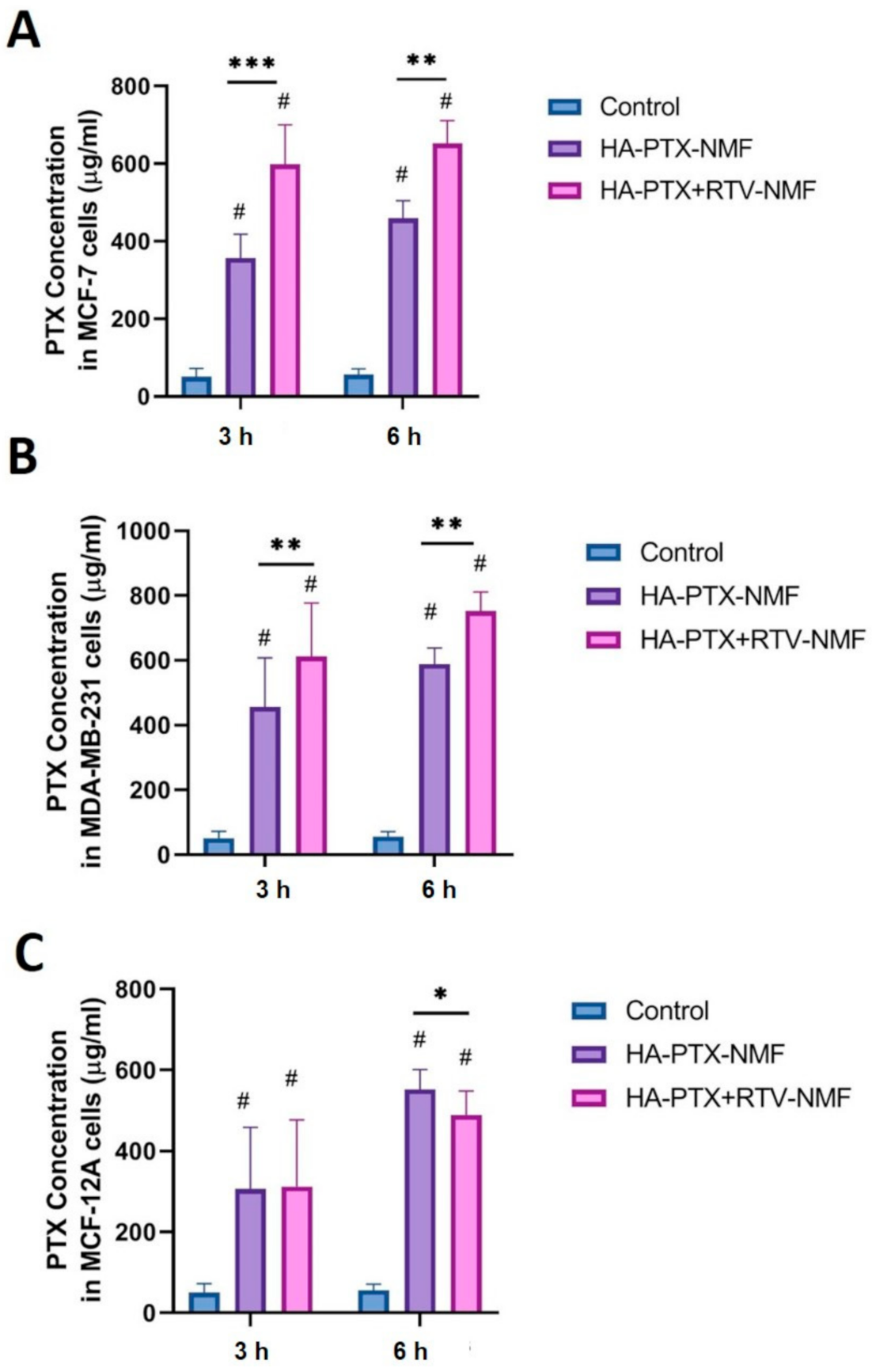
2.12. Cellular Uptake by UHPLC-MS
2.13. In Vitro Potency Determination
2.13.1. Mitochondrial Membrane Potential
2.13.2. Evaluation of Reactive Oxygen Species
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Synthesis of HA-ss-PLGA Graft Copolymer
4.4. Critical Micellar Concentration
4.5. Design of Experiment for Nanomicelles Preparation
4.6. Preparation of Nanomicellar Formulation
4.6.1. Formulation Characterization: Size, Morphology, Zeta Potential
4.6.2. Drug-Entrapment and Drug-Loading Efficiency
4.7. Nanomicellar Dilution Study
4.8. Dissolution and Drug Release
4.9. Stability Studies
4.10. Nanomicelles in Reduction Stimulated Environment
4.11. Cellular Uptake and Intracellular Distribution Study
4.11.1. FITC Labeling
4.11.2. Cellular Uptake Study by FACS
4.11.3. Intracellular Distribution Using Confocal Microscopy
4.12. Cytotoxicity Study
4.13. Cellular Uptake and P-gp Inhibition Determination by UHPLC-MS
4.14. In Vitro Potency Determination
4.14.1. Mitochondrial Membrane Potential Determination
4.14.2. Evaluation of Reactive Oxygen Species
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vadlapatla, R.K.; Patel, M.; Paturi, D.K.; Pal, D.; Mandal, A. Clinically relevant drug–drug interactions between antiretrovirals and antifungals. Expert Opin. Drug Metab. Toxicol. 2014, 10, 561–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelman, R.; Assaraf, Y.G.; Slavkin, A.; Dolev, T.; Shahar, T.; Livney, Y.D. Developing Body-Components-Based Theranostic Nanoparticles for Targeting Ovarian Cancer. Pharmaceutics 2019, 11, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiseh, M.; Kwon, D.H.; Borowsky, A.D.; Tolg, C.; Leong, H.S.; Lewis, J.D.; Turley, E.A.; Bissell, M.J. Cellular heterogeneity profiling by hyaluronan probes reveals an invasive but slow-growing breast tumor subset. Proc. Natl. Acad. Sci. USA 2014, 111, E1731–E1739. [Google Scholar] [CrossRef] [Green Version]
- Witschen, P.M.; Chaffee, T.S.; Brady, N.J.; Huggins, D.N.; Knutson, T.P.; LaRue, R.S.; Munro, S.A.; Tiegs, L.; McCarthy, J.B.; Nelson, A.C.; et al. Tumor Cell Associated Hyaluronan-CD44 Signaling Promotes Pro-Tumor Inflammation in Breast Cancer. Cancers 2020, 12, 1325. [Google Scholar] [CrossRef]
- Shim, M.S.; Kwon, Y.J. Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications. Adv. Drug Deliv. Rev. 2012, 64, 1046–1059. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, C.-F.; Iqbal, S.; Yangaf, X.; Wang, J. Responsive nanocarriers as an emerging platform for cascaded delivery of nucleic acids to cancer. Adv. Drug Deliv. Rev. 2017, 115, 98–114. [Google Scholar] [CrossRef]
- SAS, J.S.D.F. Actual by Predicted Plot. Available online: https://www.jmp.com/support/help/en/15.2/index.shtml#page/jmp/actual-by-predicted-plot-2.shtml (accessed on 20 December 2020).
- Mandal, A.; Bisht, R.; Rupenthal, I.D.; Mitra, A.K. Polymeric micelles for ocular drug delivery: From structural frameworks to recent preclinical studies. J. Control. Release 2017, 248, 96–116. [Google Scholar] [CrossRef] [Green Version]
- Gote, V.; Mandal, A.; Alshamrani, M.; Pal, D. Self-assembling tacrolimus nanomicelles for retinal drug delivery. Pharmaceutics 2020, 12, 1072. [Google Scholar] [CrossRef]
- Gote, V.; Ansong, M.; Pal, D. Prodrugs and nanomicelles to overcome ocular barriers for drug penetration. Expert Opin. Drug Metab. Toxicol. 2020, 16, 885–906. [Google Scholar] [CrossRef]
- Huang, S.; Yu, X.; Yang, L.; Song, F.; Chen, G.; Lv, Z.; Li, T.; Chen, D.; Zhu, W.; Yu, A.; et al. The efficacy of nimodipine drug delivery using mPEG-PLA micelles and mPEG-PLA/TPGS mixed micelles. Eur. J. Pharm. Sci. 2014, 63, 187–198. [Google Scholar] [CrossRef]
- Lu, Y.; Yue, Z.; Xie, J.; Wang, W.; Zhu, H.; Zhang, E.; Cao, Z. Micelles with ultralow critical micelle concentration as carriers for drug delivery. Nat. Biomed. Eng. 2018, 2, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Gaudana, R.; Ananthula, H.K.; Parenky, A.; Mitra, A.K. Ocular drug delivery. AAPS J. 2010, 12, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Cholkar, K.; Gunda, S.; Earla, R.; Pal, D.; Mitra, A.K. Nanomicellar topical aqueous drop formulation of rapamycin for back-of-the-eye delivery. AAPS PharmSciTech 2015, 16, 610–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, J. Polyoxyethylated nonionic surfactants and their applications in topical ocular drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1663–1673. [Google Scholar] [CrossRef]
- Grimaudo, M.A.; Pescina, S.; Padula, C.; Santi, P.; Concheiro, A.; Alvarez-Lorenzo, C.; Nicoli, S. Topical application of polymeric nanomicelles in ophthalmology: A review on research efforts for the noninvasive delivery of ocular therapeutics. Expert Opin. Drug Deliv. 2019, 16, 397–413. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxidative Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Liu, X.; Shao, W.; Zheng, Y.; Yao, C.; Peng, L.; Zhang, D.; Hu, X.-Y.; Wang, L. GSH-Responsive supramolecular nanoparticles constructed by beta-d-galactose-modified pillar [5]arene and camptothecin prodrug for targeted anticancer drug delivery. Chem. Commun. (Camb.) 2017, 53, 8596–8599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonafe, F.; Pazzini, C.; Marchionni, S.; Guarnieri, C.; Muscari, C. Complete disaggregation of MCF-7-derived breast tumour spheroids with very low concentrations of alpha-mangostin loaded in CD44 thioaptamer-tagged nanoparticles. Int. J. Med. Sci. 2019, 16, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, D.; Hu, J.; Ding, P.; Chen, M. Hyaluronic acid-coated pH sensitive poly (beta-amino ester) nanoparticles for co-delivery of embelin and TRAIL plasmid for triple negative breast cancer treatment. Int. J. Pharm. 2020, 573, 118637. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K. Ritonavir: A powerful boosting agent for overcoming drug resistance in cancer chemotherapy. J. Cancer Sci. Ther. 2014, 6, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Assanhou, A.G.; Li, W.; Zhang, L.; Xue, L.; Kong, L.-Y.; Sun, H.; Mo, R.; Zhang, C. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials 2015, 73, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; McCormick, B.; Wilson, K.L.; Lowes, D.A.; Colvin, L.; Torsney, C. Melatonin limits paclitaxel-induced mitochondrial dysfunction in vitro and protects against paclitaxel-induced neuropathic pain in the rat. J. Pineal Res. 2017, 63, e12444. [Google Scholar] [CrossRef] [Green Version]
- Berard, P.V.; Mulfinger, H.; Gambarelli-Mouillac, N. Cysteine. Results of its use in instillations for treatment of corneal diseases. Bull. Soc. Ophtalmol. Fr. 1976, 76, 219–222. [Google Scholar]
- Pustylnikov, S.; Costabile, F.; Beghi, S.; Facciabene, A. Targeting mitochondria in cancer: Current concepts and immunotherapy approaches. Transl. Res. 2018, 202, 35–51. [Google Scholar] [CrossRef]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Bu, H.; He, X.; Zhang, Z.; Yin, Q.; Yu, H.; Li, Y. A TPGS-incorporating nanoemulsion of paclitaxel circumvents drug resistance in breast cancer. Int. J. Pharm. 2014, 471, 206–213. [Google Scholar] [CrossRef]
- Aghaei, M.; Shahali, A.; Ghanadian, M.; Jafari, S.M. Mitochondrial and caspase pathways are involved in the induction of apoptosis by nardosinen in MCF-7 breast cancer cell line. Res. Pharm. Sci. 2018, 13, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K.-M.; Poncet, D.; Kroemer, G. Chemotherapy: Targeting the mitochondrial cell death pathway. Oncogene 2002, 21, 8786–8803. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Li, Y.-B.; Yao, H.-J.; Ju, R.-J.; Zhang, Y.; Li, R.-J.; Yu, Y.; Zhang, L.; Lu, W.-L. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyeth-ylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials 2011, 32, 5673–5687. [Google Scholar] [CrossRef]
- Kanamala, M.; Wilson, W.R.; Yang, M.; Palmer, B.D.; Wu, Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials 2016, 85, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Holzhütter, H.-G.; Bogyo, M.; Kairies, N.; Groll, M.; De Giuli, R.; Emch, S.; Groettrup, M. How an inhibitor of the HIV-I protease modulates proteasome activity. J. Biol. Chem. 1999, 274, 35734–35740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mitra, A.K. Mechanisms of drug resistance in cancer chemotherapy: Coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr. Pharm. Des. 2013, 19, 7126–7140. [Google Scholar] [CrossRef] [PubMed]
- Amawi, H.; Sim, H.-M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-mediated multidrug-resistant cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef]
- Lee, G.-Y.; Lee, J.-S.; Son, C.-G.; Lee, N. Combating drug resistance in colorectal cancer using herbal medicines. Chin. J. Integr. Med. 2020, 1–10. [Google Scholar] [CrossRef]
- Hems, D. Biochemical aspects of renal ammonia formation in metabolic acidosis. Enzyme 1975, 20, 359–380. [Google Scholar] [CrossRef]
- Zhang, F.L.; Cao, J.-L.; Xie, H.-Y.; Sun, R.; Yang, L.-F.; Shao, Z.-M.; Li, D.-Q. Cancer-associated MORC2-mutant M276I regulates an hnRNPM-mediated CD44 splicing switch to promote invasion and metastasis in triple-negative breast cancer. Cancer Res. 2018, 78, 5780–5792. [Google Scholar] [CrossRef] [Green Version]
- Asano, M.; Tanaka, S.; Sakaguchi, M. Effects of normothermic microwave irradiation on CD44+/CD24‒ in breast cancer MDA-MB-231 and MCF-7 cell lines. Biosci. Biotechnol. Biochem. 2020, 84, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Al-Othman, N.; Alhendi, A.; Ihbaisha, M.; Barahmeh, M.; Alqaraleh, M.; Al-Momany, B.Z. Role of CD44 in breast cancer. Breast Dis. 2020, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Madaan, A.; Singh, P.; Awasthi, A.; Verma, R.; Singh, A.T.; Jaggi, M.; Mishra, S.K.; Kulkarni, S.; Kulkarni, H. Efficiency and mechanism of intracellular paclitaxel delivery by novel nanopolymer-based tumor-targeted delivery system, Nanoxel(TM). Clin. Transl. Oncol. 2013, 15, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Cholkar, K.; Khurana, V.; Shah, A.; Agrahari, V.; Bisht, R.; Pal, D.; Mitra, A.K. Topical formulation of self-assembled antiviral prodrug nanomicelles for targeted retinal delivery. Mol. Pharm. 2017, 14, 2056–2069. [Google Scholar] [CrossRef] [PubMed]
- Ngatuni, M.J.; Trinh, H.M.; Pal, D.; Mitra, A.K. Novel Random Triblock Copolymers for Sustained Delivery of Macromolecules for the Treatment of Ocular Diseases. AAPS PharmSciTech 2018, 19, 3871–3885. [Google Scholar] [CrossRef] [PubMed]
- Zabetakis, D.; Olson, M.A.; Anderson, G.P.; Legler, P.M.; Goldman, E.R. Evaluation of disulfide bond position to enhance the thermal stability of a highly stable single domain antibody. PLoS ONE 2014, 9, e115405. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Huai, J.; Chen, X.; Yang, Y.; Zhang, X.; Gan, Y.; Wang, G.; Gu, X.; Li, J. Intracellular delivery and antitumor effects of a redox-responsive polymeric paclitaxel conjugate based on hyaluronic acid. Acta Biomater. 2015, 26, 274–285. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Roy, R.; Ventura, J.; Thayumanavan, S. Redox-sensitive disassembly of amphiphilic copolymer based micelles. Langmuir 2010, 26, 7086–7092. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Name | Coded Design | Uncoded Design | ||||
|---|---|---|---|---|---|---|
| X1 | X2 | X3 | X1 = Sonication Time (min) | X2 = HA-ss-PLGA (wt%) | X3 = Vit E-TPGS (wt%) | |
| F1 | 0 | 0 | 0 | 22.5 | 2 | 4 |
| F2 | 0 | 0 | 0 | 22.5 | 2 | 4 |
| F3 | − | − | + | 20 | 0.5 | 5 |
| F4 | − | + | + | 20 | 3.5 | 5 |
| F5 | 0 | 0 | 0 | 22.5 | 2 | 4 |
| F6 | + | − | + | 25 | 0.5 | 5 |
| F7 | + | − | − | 25 | 0.5 | 3 |
| F8 | − | − | − | 20 | 0.5 | 3 |
| F9 | + | + | + | 25 | 3.5 | 5 |
| F10 | − | + | − | 20 | 3.5 | 3 |
| F11 | + | + | − | 25 | 3.5 | 3 |
| Formulation | Pattern | Sonication Time (min) | HA−PLGA (wt%) | Vit E TPGS(wt%) | Size (nm) | PDI | Zeta (mV) | PTX | RTV | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EE% | LE% | EE% | LE% | ||||||||
| F1 | (0)(0)(0) | 22.5 | 2 | 4 | 144.0 | 0.2 | 0.2 | 96.5 | 7.4 | 95.5 | 1.9 |
| F2 | (0)(0)(0) | 22.5 | 2 | 4 | 146.0 | 0.2 | 0.2 | 95.7 | 7.2 | 96.9 | 1.8 |
| F3 | (−)(−)(+) | 20 | 0.5 | 5 | 168.8 | 0.5 | 0.0 | 83.0 | 6.9 | 84.2 | 1.9 |
| F4 | (−)(+)(+) | 20 | 3.5 | 5 | 256.2 | 0.6 | 0.0 | 88.9 | 4.9 | 90.0 | 1.3 |
| F5 | (0)(0)(0) | 22.5 | 2 | 4 | 142.5 | 0.2 | 0.1 | 95.4 | 7.2 | 96.0 | 1.8 |
| F6 | (+)(−)(+) | 25 | 0.5 | 5 | 159.6 | 0.8 | 0.2 | 85.8 | 7.1 | 86.7 | 1.6 |
| F7 | (+)(−)(−) | 25 | 0.5 | 3 | 204.3 | 0.3 | 0.0 | 73.7 | 9.2 | 93.8 | 3.2 |
| F8 | (−)(−)(−) | 20 | 0.5 | 3 | 179.7 | 0.3 | 0.0 | 74.0 | 9.3 | 93.4 | 3.2 |
| F9 | (+)(+)(+) | 25 | 3.5 | 5 | 189.9 | 0.6 | 0.0 | 88.4 | 5.0 | 89.2 | 1.3 |
| F10 | (−)(+)(−) | 20 | 3.5 | 3 | 249.8 | 0.5 | 0.2 | 76.9 | 5.5 | 78.7 | 1.5 |
| F11 | (+)(+)(−) | 20 | 3.5 | 3 | 225.0 | 0.4 | 0.2 | 75.5 | 5.4 | 75.9 | 1.4 |
| Dilution Factor | Hydrodynamic Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 °C | R.T. | 37 °C | 4 °C | R.T. | 37 °C | 4 °C | R.T. | 37 °C | |
| 0 | 144.1 | 144.2 | 156.3 | 0.25 | 0.183 | 0.26 | 0.46 | 0.522 | 0.53 |
| 10 | 144.6 | 144.9 | 159.6 | 0.21 | 0.251 | 0.29 | 0.56 | 0.651 | 0.95 |
| 50 | 144.8 | 145.2 | 165.3 | 0.26 | 0.259 | 0.21 | 0.85 | 1.016 | 1.63 |
| 100 | 149.6 | 147.5 | 169.7 | 0.25 | 0.322 | 0.31 | 0.89 | 1.13 | 1.46 |
| 150 | 152.0 | 150.3 | 170.3 | 0.29 | 0.468 | 0.34 | 1.26 | 1.523 | 1.62 |
| 200 | 154.9 | 155.4 | 176.4 | 0.35 | 0.672 | 0.5 | 1.65 | 1.92 | 2.30 |
| Time Points | Hydrodynamic Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 °C | 25 °C | 40 °C | 4 °C | 25 °C | 40 °C | 4 °C | 25 °C | 40 °C | |
| Time 0 | 145.1 | 144.2 | 151.3 | 0.13 | 0.16 | 0.29 | 0.96 | 0.722 | 1.53 |
| Day 3 | 149.6 | 165.9 | 397.6 | 0.22 | 0.25 | 0.56 | 1.6 | 1.651 | 4.95 |
| Day 7 | 156.8 | 175.4 | 572.3 | 0.26 | 0.39 | 0.61 | 1.25 | 2.160 | 5.63 |
| Time Points | Hydrodynamic Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) |
|---|---|---|---|
| Time 0 | 149.5 | 0.26 | 8.96 |
| Day 3 | 163.6 | 0.45 | 12.3 |
| Day 7 | 262.3 | 0.51 | 16.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gote, V.; Sharma, A.D.; Pal, D. Hyaluronic Acid-Targeted Stimuli-Sensitive Nanomicelles Co-Encapsulating Paclitaxel and Ritonavir to Overcome Multi-Drug Resistance in Metastatic Breast Cancer and Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1257. https://doi.org/10.3390/ijms22031257
Gote V, Sharma AD, Pal D. Hyaluronic Acid-Targeted Stimuli-Sensitive Nanomicelles Co-Encapsulating Paclitaxel and Ritonavir to Overcome Multi-Drug Resistance in Metastatic Breast Cancer and Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(3):1257. https://doi.org/10.3390/ijms22031257
Chicago/Turabian StyleGote, Vrinda, Amar Deep Sharma, and Dhananjay Pal. 2021. "Hyaluronic Acid-Targeted Stimuli-Sensitive Nanomicelles Co-Encapsulating Paclitaxel and Ritonavir to Overcome Multi-Drug Resistance in Metastatic Breast Cancer and Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 3: 1257. https://doi.org/10.3390/ijms22031257
APA StyleGote, V., Sharma, A. D., & Pal, D. (2021). Hyaluronic Acid-Targeted Stimuli-Sensitive Nanomicelles Co-Encapsulating Paclitaxel and Ritonavir to Overcome Multi-Drug Resistance in Metastatic Breast Cancer and Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences, 22(3), 1257. https://doi.org/10.3390/ijms22031257