
The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia



Abstract
:1. Introduction
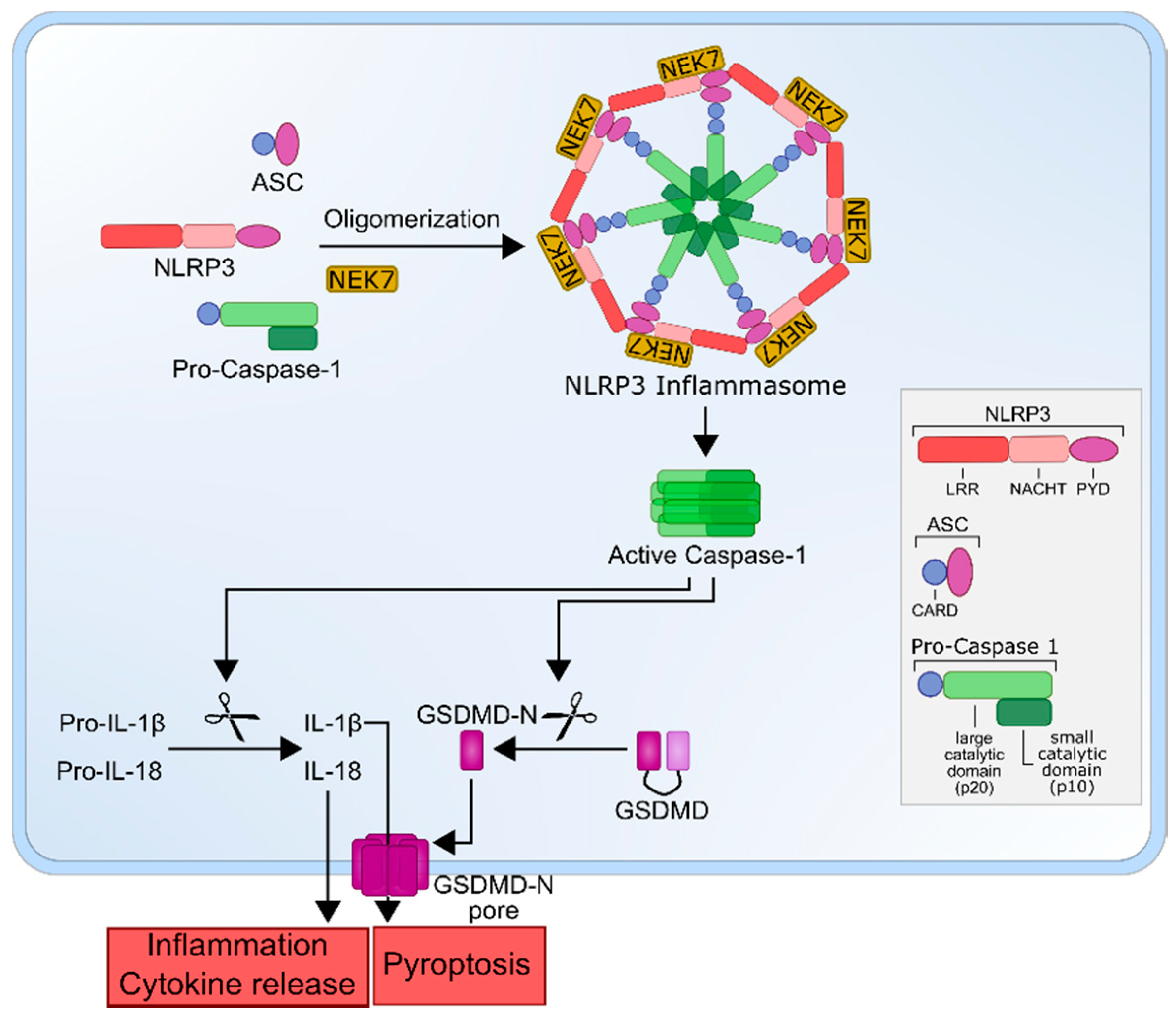
2. The NLRP3 Inflammasome
3. The Role of the NLRP3 Inflammasome in Different Types of Leukemia
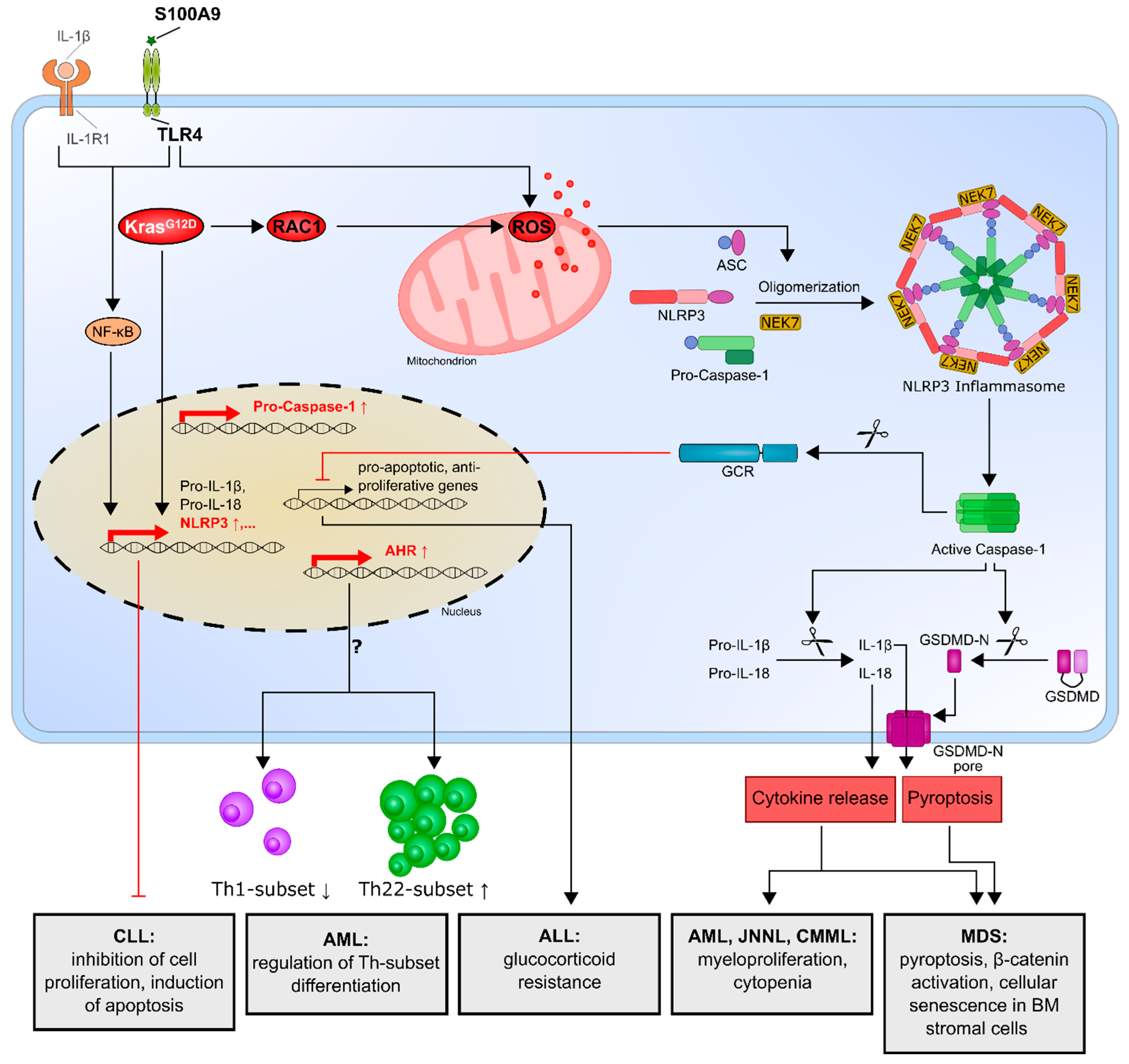
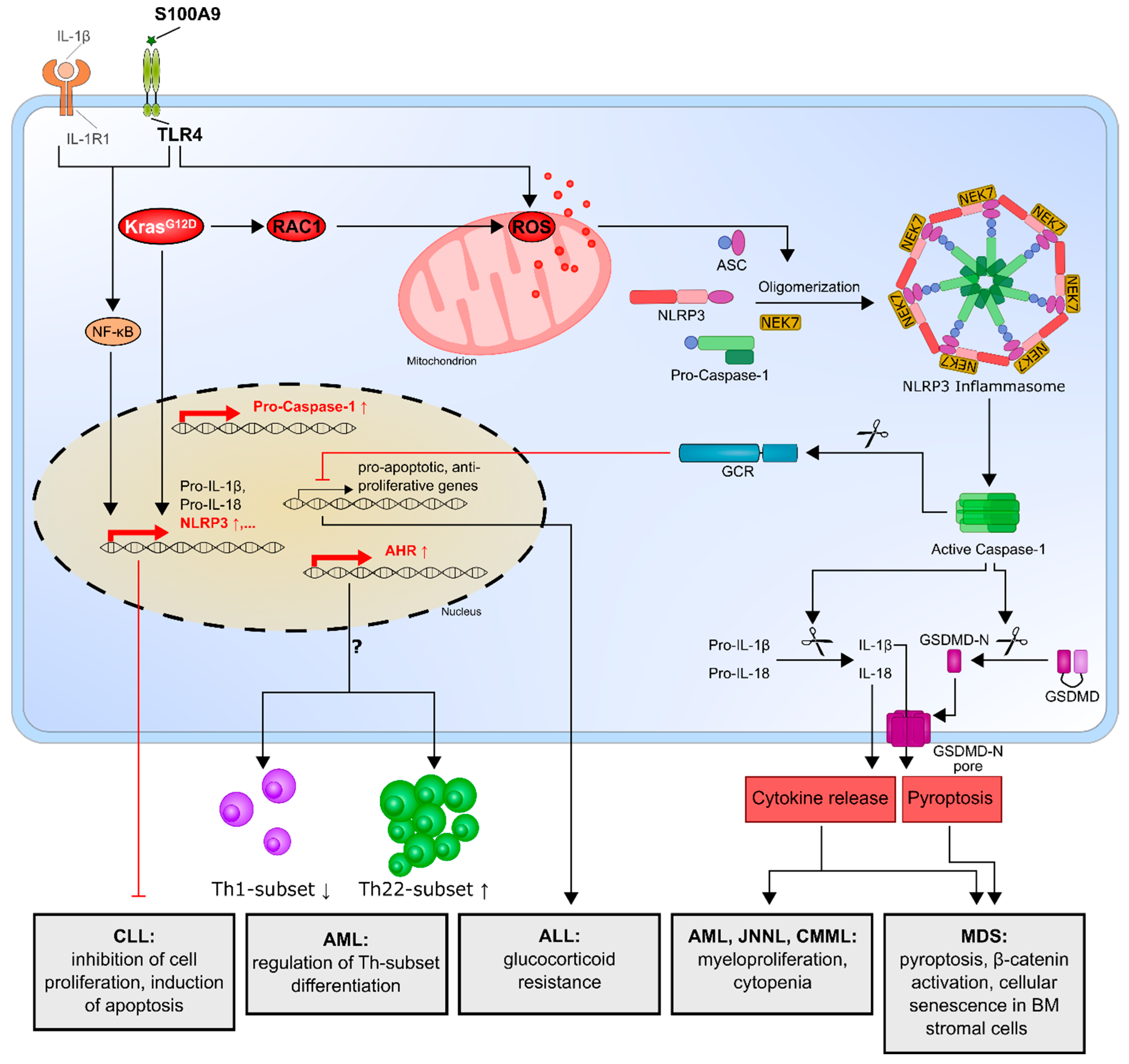
3.1. Myelodysplastic Syndrome (MDS)
3.2. Acute Myeloid Leukemia (AML)
3.3. Acute Lymphocytic Leukemia (ALL)
3.4. Chronic Lymphocytic Leukemia (CLL)
4. The NLRP3 Inflammasome as a Therapeutic Target
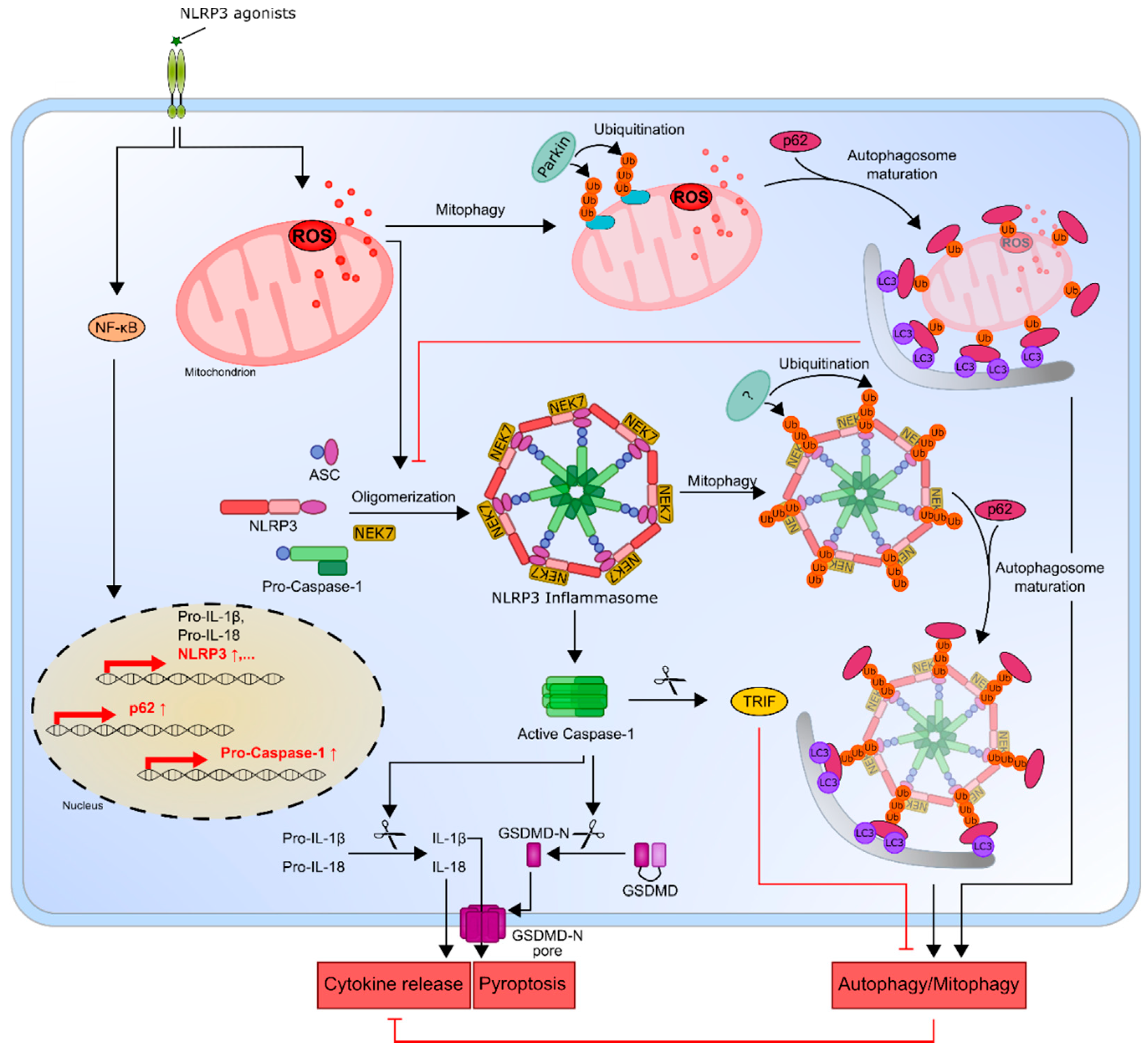
5. The NLRP3 Inflammasome and Its Connection to Autophagy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J.; et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2016, 2, 16010. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.; Dalla-Favera, R. Chronic lymphocytic leukaemia: From genetics to treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute. Cancer Stat Facts: Leukemia. Available online: https://seer.cancer.gov/statfacts/html/leuks.html (accessed on 4 November 2020).
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Landskron, G.; de la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; el Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The critical role of inflammation in the pathogenesis and progression of myeloid malignancies. Cancers 2018, 10, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Ponzetta, A.; Inforzato, A.; Jaillon, S. Innate immunity, inflammation and tumour progression: Double-edged swords. J. Intern. Med. 2019, 285, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Madej, M.P.; Topfer, E.; Boraschi, D.; Italiani, P. Different regulation of interleukin-1 production and activity in monocytes and macrophages: Innate memory as an endogenous mechanism of IL-1 inhibition. Front. Pharmacol. 2017, 8, 335. [Google Scholar] [CrossRef]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1beta as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 beta-A friend or foe in malignancies? Int. J. Mol. Sci 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, F.; Rubartelli, A.; Aldinucci, D.; Sitia, R.; Torcia, M.; Shaw, A.; di Guglielmo, R. Interleukin 1 as an autocrine growth factor for acute myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 1989, 86, 2369–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, A.; Edwards, D.K.T.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. 2017, 18, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Perez-Figueroa, E.; Sanchez-Cuaxospa, M.; Martinez-Soto, K.A.; Sanchez-Zauco, N.; Medina-Sanson, A.; Jimenez-Hernandez, E.; Torres-Nava, J.R.; Felix-Castro, J.M.; Gomez, A.; Ortega, E.; et al. Strong inflammatory response and Th1-polarization profile in children with acute lymphoblastic leukemia without apparent infection. Oncol. Rep. 2016, 35, 2699–2706. [Google Scholar] [CrossRef]
- Vilchis-Ordonez, A.; Contreras-Quiroz, A.; Vadillo, E.; Dorantes-Acosta, E.; Reyes-Lopez, A.; del Prado, H.M.Q.-N.; Venegas-Vazquez, J.; Mayani, H.; Ortiz-Navarrete, V.; Lopez-Martinez, B.; et al. Bone marrow cells in acute lymphoblastic leukemia create a proinflammatory microenvironment influencing normal hematopoietic differentiation fates. Biomed. Res. Int. 2015, 2015, 386165. [Google Scholar] [CrossRef]
- Agerstam, H.; Hansen, N.; von Palffy, S.; Sanden, C.; Reckzeh, K.; Karlsson, C.; Lilljebjorn, H.; Landberg, N.; Askmyr, M.; Hogberg, C.; et al. IL1RAP antibodies block IL-1-induced expansion of candidate CML stem cells and mediate cell killing in xenograft models. Blood 2016, 128, 2683–2693. [Google Scholar] [CrossRef]
- Agerstam, H.; Karlsson, C.; Hansen, N.; Sanden, C.; Askmyr, M.; von Palffy, S.; Hogberg, C.; Rissler, M.; Wunderlich, M.; Juliusson, G.; et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2015, 112, 10786–10791. [Google Scholar] [CrossRef] [Green Version]
- Ennas, M.G.; Moore, P.S.; Zucca, M.; Angelucci, E.; Cabras, M.G.; Melis, M.; Gabbas, A.; Serpe, R.; Madeddu, C.; Scarpa, A.; et al. Interleukin-1B (IL1B) and interleukin-6 (IL6) gene polymorphisms are associated with risk of chronic lymphocytic leukaemia. Hematol. Oncol. 2008, 26, 98–103. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; di Paola, R. Focus on the role of NLRP3 inflammasome in diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Zeiser, R. NLRP3 inflammasome activation in cancer: A double-edged sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kanneganti, T.D. The inflammasome: Firing up innate immunity. Immunol. Rev. 2015, 265, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.K.; Kucia, M. The Nlrp3 inflammasome as a “rising star” in studies of normal and malignant hematopoiesis. Leukemia 2020, 34, 1512–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [Green Version]
- Hafner-Bratkovic, I.; Susjan, P.; Lainscek, D.; Tapia-Abellan, A.; Cerovic, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrin, P.; Jerala, R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018, 9, 5182. [Google Scholar] [CrossRef] [Green Version]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef] [Green Version]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Li, C.C.; Di, B.; Xu, L.L. Recent advances in the NEK7-licensed NLRP3 inflammasome activation: Mechanisms, role in diseases and related inhibitors. J. Autoimmun. 2020, 113, 102515. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Eigenbrod, T.; Nunez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K (+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10, 346. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.D.; Rehman, J.; et al. The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 2018, 49, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, Z.; Wang, C.; Yang, R.; Rathkey, J.K.; Pinkard, O.W.; Shi, W.; Chen, Y.; Dubyak, G.R.; Abbott, D.W.; et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, 6792–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Wang, X.; Zheng, Y.; Jiang, J.; Hu, J. What role does pyroptosis play in microbial infection? J. Cell Physiol. 2019, 234, 7885–7892. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front. Immunol. 2017, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [Green Version]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglie, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hua, M.; Wang, S.; Yu, J.; Chen, C.; Zhao, X.; Zhang, C.; Zhong, C.; Wang, R.; He, N.; et al. Genetic polymorphisms of IL-18 rs1946518 and IL-1beta rs16944 are associated with prognosis and survival of acute myeloid leukemia. Inflamm. Res. 2017, 66, 249–258. [Google Scholar] [CrossRef]
- Zhang, C.; Han, F.; Yu, J.; Hu, X.; Hua, M.; Zhong, C.; Wang, R.; Zhao, X.; Shi, Y.; Ji, C.; et al. Investigation of NF-kappaB-94ins/del ATTG and CARD8 (rs2043211) gene polymorphism in acute lymphoblastic leukemia. Front. Endocrinol. 2019, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Yu, J.; Yan, S.; Zhao, X.; Chen, C.; Zhou, Y.; Zhao, X.; Hua, M.; Wang, R.; Zhang, C.; et al. The genetic polymorphism and expression profiles of NLRP3 inflammasome in patients with chronic myeloid leukemia. Hum. Immunol. 2018, 79, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; He, N.; Li, P.; Zhang, C.; Yu, J.; Hua, M.; Ji, C.; Ma, D. Polymorphisms of Interlukin-1beta rs16944 confer susceptibility to myelodysplastic syndromes. Life Sci. 2016, 165, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Cluzeau, T.; Basiorka, A.A.; List, A. Unraveling the pathogenesis of MDS: The NLRP3 inflammasome and pyroptosis drive the MDS phenotype. Front. Oncol. 2016, 6, 151. [Google Scholar] [CrossRef]
- Shi, L.; Zhao, Y.; Fei, C.; Guo, J.; Jia, Y.; Wu, D.; Wu, L.; Chang, C. Cellular senescence induced by S100A9 in mesenchymal stromal cells through NLRP3 inflammasome activation. Aging 2019, 11, 9626–9642. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; de Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lubbert, M.; et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Zhang, C.; Hua, M.; Wang, M.; Chen, P.; Ma, D. Aberrant NLRP3 inflammasome associated with aryl hydrocarbon receptor potentially contributes to the imbalance of T-helper cells in patients with acute myeloid leukemia. Oncol. Lett. 2017, 14, 7031–7044. [Google Scholar] [CrossRef] [Green Version]
- Paugh, S.W.; Bonten, E.J.; Savic, D.; Ramsey, L.B.; Thierfelder, W.E.; Gurung, P.; Malireddi, R.K.; Actis, M.; Mayasundari, A.; Min, J.; et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 2015, 47, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Salaro, E.; Rambaldi, A.; Falzoni, S.; Amoroso, F.S.; Franceschini, A.; Sarti, A.C.; Bonora, M.; Cavazzini, F.; Rigolin, G.M.; Ciccone, M.; et al. Involvement of the P2X7-NLRP3 axis in leukemic cell proliferation and death. Sci. Rep. 2016, 6, 26280. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, A.A. Myelodysplastic syndrome from theoretical review to clinical application view. Oncol. Rev. 2018, 12, 397. [Google Scholar] [CrossRef]
- Azizi, G.; Pouyani, M.R.; Navabi, S.S.; Yazdani, R.; Kiaee, F.; Mirshafiey, A. The newly identified T helper 22 cells lodge in leukemia. Int. J. Hematol. Oncol. Stem Cell Res. 2015, 9, 143–154. [Google Scholar] [PubMed]
- Schmidt, S.; Rainer, J.; Ploner, C.; Presul, E.; Riml, S.; Kofler, R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: Molecular mechanisms and clinical relevance. Cell Death Differ. 2004, 11, S45–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploner, C.; Schmidt, S.; Presul, E.; Renner, K.; Schrocksnadel, K.; Rainer, J.; Riml, S.; Kofler, R. Glucocorticoid-induced apoptosis and glucocorticoid resistance in acute lymphoblastic leukemia. J. Steroid Biochem. Mol. Biol. 2005, 93, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Clarisse, D.; Offner, F.; de Bosscher, K. Latest perspectives on glucocorticoid-induced apoptosis and resistance in lymphoid malignancies. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188430. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.J.; Hu, C.G.; Zhu, Z.M.; Luo, H.L. Effect of P2X7 receptor on tumorigenesis and its pharmacological properties. Biomed. Pharmacother. 2020, 125, 109844. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, A.; Lv, J.; Zhang, Q.; Ran, Y.; Wei, C.; Wu, J. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur. J. Med. Chem. 2020, 185, 111822. [Google Scholar] [CrossRef]
- Gouravani, M.; Khalili, N.; Razi, S.; Keshavarz-Fathi, M.; Khalili, N.; Rezaei, N. The NLRP3 inflammasome: A therapeutic target for inflammation-associated cancers. Expert Rev. Clin. Immunol. 2020, 16, 175–187. [Google Scholar] [CrossRef]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef] [Green Version]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [Green Version]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, M. IL-1 trap go-ahead. Nat. Biotechnol. 2008, 26, 485. [Google Scholar] [CrossRef] [PubMed]
- Askmyr, M.; Agerstam, H.; Hansen, N.; Gordon, S.; Arvanitakis, A.; Rissler, M.; Juliusson, G.; Richter, J.; Jaras, M.; Fioretos, T. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 2013, 121, 3709–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacha, T.; Saglio, G. Nilotinib in the treatment of chronic myeloid leukemia. Future Oncol. 2019, 15, 953–965. [Google Scholar] [CrossRef]
- Zhang, B.; Chu, S.; Agarwal, P.; Campbell, V.L.; Hopcroft, L.; Jorgensen, H.G.; Lin, A.; Gaal, K.; Holyoake, T.L.; Bhatia, R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 2016, 128, 2671–2682. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, C.; Araiz, A.R.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [Green Version]
- Tapia-Abellan, A.; Angosto-Bazarra, D.; Martinez-Banaclocha, H.; de Torre-Minguela, C.; Ceron-Carrasco, J.P.; Perez-Sanchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Ismael, S.; Nasoohi, S.; Ishrat, T. MCC950, the selective inhibitor of nucleotide oligomerization domain-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J. Neurotrauma 2018, 35, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Meng, X.; Ye, T.; Xie, W.; Sun, G.; Sun, X. Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates diabetic encephalopathy in db/db mice. Molecules 2018, 23, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Perera, A.P.; Fernando, R.; Shinde, T.; Gundamaraju, R.; Southam, B.; Sohal, S.S.; Robertson, A.A.B.; Schroder, K.; Kunde, D.; Eri, R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep. 2018, 8, 8618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Koenders, M.I.; Azam, T.; Tengesdal, I.W.; Powers, N.; de Graaf, D.M.; Dinarello, C.A.; Joosten, L.A.B. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res. Ther. 2018, 20, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Fernandez, A.; Skouras, D.B.; Dinarello, C.A.; Lopez-Vales, R. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front. Immunol. 2019, 10, 2578. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, C.; Zhang, Y. Therapeutic effect of Rabdosia rubescens aqueous extract on chronic pharyngitis and its safety. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011, 36, 170–173. [Google Scholar] [CrossRef]
- Kuo, L.M.; Kuo, C.Y.; Lin, C.Y.; Hung, M.F.; Shen, J.J.; Hwang, T.L. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules 2014, 19, 3327–3344. [Google Scholar] [CrossRef]
- Yang, J.; Jiang, H.; Wang, C.; Yang, B.; Zhao, L.; Hu, D.; Qiu, G.; Dong, X.; Xiao, B. Oridonin triggers apoptosis in colorectal carcinoma cells and suppression of microRNA-32 expression augments oridonin-mediated apoptotic effects. Biomed. Pharmacother. 2015, 72, 125–134. [Google Scholar] [CrossRef]
- Ding, Y.; Ding, C.; Ye, N.; Liu, Z.; Wold, E.A.; Chen, H.; Wild, C.; Shen, Q.; Zhou, J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016, 122, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.; Moore, R.D. Disulfiram treatment of alcoholism. Am. J. Med. 1990, 88, 647–655. [Google Scholar] [CrossRef]
- Xu, B.; Shi, P.; Fombon, I.S.; Zhang, Y.; Huang, F.; Wang, W.; Zhou, S. Disulfiram/copper complex activated JNK/c-jun pathway and sensitized cytotoxicity of doxorubicin in doxorubicin resistant leukemia HL60 cells. Blood Cells Mol. Dis. 2011, 47, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; Chen, F.; Dong, H.; Shi, P.; Yao, Y.; Zhang, Y.; Li, R.; Wang, S.; Li, P.; Wang, W.; et al. Disulfiram targeting lymphoid malignant cell lines via ROS-JNK activation as well as Nrf2 and NF-kB pathway inhibition. J. Transl. Med. 2014, 12, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Xie, J.; Hou, R.; Chen, X.; Xu, Z.; Tan, Y.; Ren, F.; Zhang, Y.; Xu, J.; Chang, J.; et al. Disulfiram/cytarabine eradicates a subset of acute myeloid leukemia stem cells with high aldehyde dehydrogenase expression. Leuk. Res. 2020, 92, 106351. [Google Scholar] [CrossRef]
- Hassani, S.; Ghaffari, P.; Chahardouli, B.; Alimoghaddam, K.; Ghavamzadeh, A.; Alizadeh, S.; Ghaffari, S.H. Disulfiram/copper causes ROS levels alteration, cell cycle inhibition, and apoptosis in acute myeloid leukaemia cell lines with modulation in the expression of related genes. Biomed. Pharmacother. 2018, 99, 561–569. [Google Scholar] [CrossRef]
- Deng, M.; Jiang, Z.; Li, Y.; Zhou, Y.; Li, J.; Wang, X.; Yao, Y.; Wang, W.; Li, P.; Xu, B. Effective elimination of adult B-lineage acute lymphoblastic leukemia by disulfiram/copper complex in vitro and in vivo in patient-derived xenograft models. Oncotarget 2016, 7, 82200–82212. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Yoon, S.O.; Lee, H.J.; Oh, J.Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFkappaB pathways in autophagy- and p62-dependent manners. Oncotarget 2017, 8, 40817–40831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Yoon, J.H.; Ryu, J.H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wang, Y.; Long, Z.; He, G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. 2019, 51, 1087–1095. [Google Scholar] [CrossRef]
- Lai, M.; Yao, H.; Shah, S.Z.A.; Wu, W.; Wang, D.; Zhao, Y.; Wang, L.; Zhou, X.; Zhao, D.; Yang, L. The NLRP3-caspase 1 inflammasome negatively regulates autophagy via TLR4-TRIF in prion peptide-infected microglia. Front. Aging Neurosci. 2018, 10, 116. [Google Scholar] [CrossRef]
- Jabir, M.S.; Ritchie, N.D.; Li, D.; Bayes, H.K.; Tourlomousis, P.; Puleston, D.; Lupton, A.; Hopkins, L.; Simon, A.K.; Bryant, C.; et al. Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and beta-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe 2014, 15, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Tholken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Surun, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Wang, L.; Cheng, S.; Wang, Y.; Zhao, W. Autophagy and leukemia. Adv. Exp. Med. Biol. 2020, 1207, 601–613. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic modulation of autophagy in leukaemia and lymphoma. Cells 2019, 8, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Type of Hematologic Malignancy | Key Findings | Reference |
|---|---|---|
| MDS | NLRP3 inflammasome activation in MDS disorders is responsible for the key biological features of MDS, which drive pyroptotic cell death and β-catenin activation. | [74,75] |
| Cellular senescence in bone marrow stromal cells from MDS patients is induced by increased S100A9 expression through TLR4, NLRP3 inflammasome activation and IL-1β secretion. | [76] | |
| AML, CMML, JNNL | Oncogenic KrasG12D mutation activates the KRAS/RAC1/ROS/NLRP3/IL-1β axis and promotes myeloproliferation and cytopenia. | [77] |
| AML | Enhanced NLRP3 expression correlates with an increased aryl hydrocarbon receptor and might influence T-helper cell differentiation. | [78] |
| ALL | Overexpression of NLRP3 and caspase-1 is responsible for glucocorticoid resistance through the cleavage of the glucocorticoid receptor by caspase-1. | [79] |
| CLL | NLRP3 negatively regulates the progression of CLL by promoting the expression of P2X7R, while NLRP3 overexpression inhibits cell proliferation and survival. | [80] |
| Inhibitor | Inhibition Mechanism | Reference |
|---|---|---|
| MCC950 | Binds Walker B motif of the NLRP3 NACHT domain; NACHT ATPase inhibitor | [98,99] |
| CY-09 | Binds Walker A motif of the NLRP3 NACHT domain; NACHT ATPase inhibitor | [100] |
| OLT1177 | NACHT ATPase inhibitor | [101] |
| Tranilast | Binds the NLRP3 NACHT domain and inhibits NLRP3–NLRP3 interaction | [102] |
| Oridonin | Binds irreversibly to NLRP3 Cys279 and inhibits NLRP3–NEK7 interaction | [103] |
| Disulfiram | Blocks gasdermin D pore formation and inhibits pyroptosis and cytokine release | [104] |
| Necrosulfonamide (NSA) | Binds to gasdermin D and prevents pyroptosis | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urwanisch, L.; Luciano, M.; Horejs-Hoeck, J. The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia. Int. J. Mol. Sci. 2021, 22, 1271. https://doi.org/10.3390/ijms22031271
Urwanisch L, Luciano M, Horejs-Hoeck J. The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia. International Journal of Molecular Sciences. 2021; 22(3):1271. https://doi.org/10.3390/ijms22031271
Chicago/Turabian StyleUrwanisch, Laura, Michela Luciano, and Jutta Horejs-Hoeck. 2021. "The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia" International Journal of Molecular Sciences 22, no. 3: 1271. https://doi.org/10.3390/ijms22031271
APA StyleUrwanisch, L., Luciano, M., & Horejs-Hoeck, J. (2021). The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia. International Journal of Molecular Sciences, 22(3), 1271. https://doi.org/10.3390/ijms22031271