Reorganization of Metabolism during Cardiomyogenesis Implies Time-Specific Signaling Pathway Regulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Exploration of Cellular Metabolism during Cardiomyogenic Differentiation
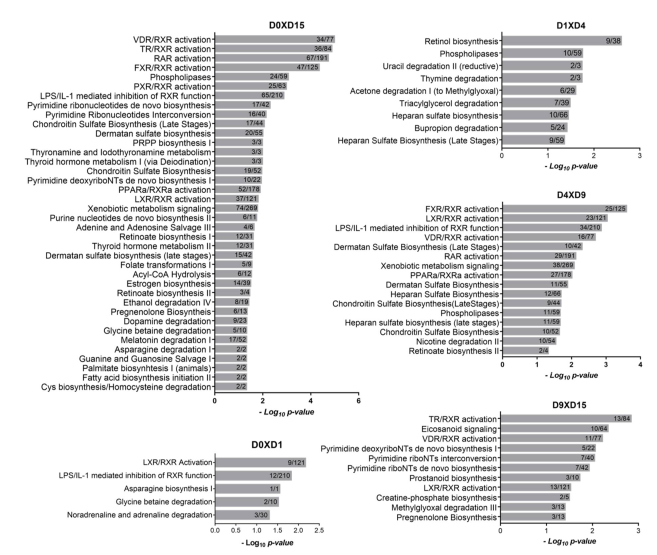
2.1.1. Metabolism-Related Canonical Pathways
2.1.2. Energy Metabolism Changes between hESCs and hESC-CM
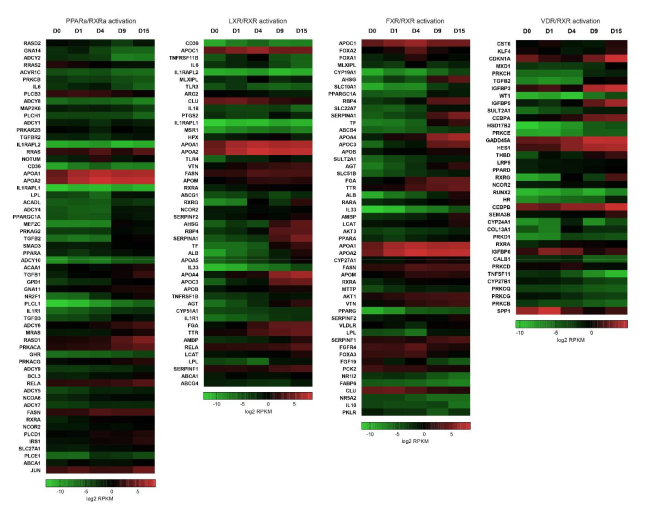
2.2. RXR-Dependent Signaling Pathways Are Regulated during Cardiomyogenesis
2.2.1. Heterodimers of RXR with PPAR, LXR and FXR Can Modulate Lipid Homeostasis
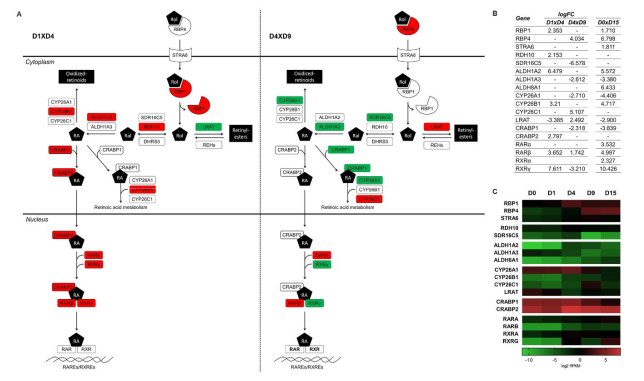
2.2.2. Stage-Specific Regulation of Retinoic Acid Metabolism
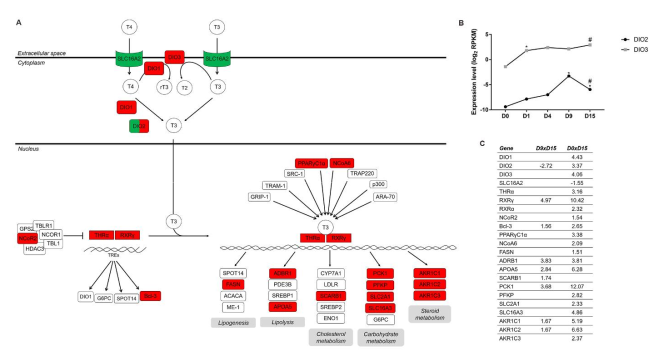
2.2.3. TH Metabolism and Signaling Influence Downstream Pathways Regulating Cardiomyogenesis
2.3. Additional Lipid Metabolism-Related Pathways Are Enriched during Cardiac Differentiation
2.4. Heparan Sulfate Biosynthesis: Modification Enzymes and Cell Fate Regulation
2.5. Biological Functions Related to Metabolism are Stage-Specifically Regulated
3. Concluding Remarks
4. Materials and Methods
4.1. Cardiomyocyte Differentiation, Polysome Profiling and Sequencing
4.2. Bioinformatic RNA-seq Analysis
4.3. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| hESC | Human embryonic stem cells |
| hESC-CMs | Human embryonic stem cells-derived cardiomyocytes |
| EB | Embryoid body |
| CP | Cardiac progenitor |
| CM | Cardiomyocyte |
| ECM | Extracellular matrix |
| RPKM | Reads per kilobase per million mapped reads |
| DEG | Differentially expressed genes |
| IPA | Ingenuity pathways analysis |
| FC | Fold change |
| FDR | False discovery rate |
References
- Zhang, J.; Zhao, J.; Dahan, P.; Lu, V.; Zhang, C.; Li, H.; Teitell, M.A. Metabolism in Pluripotent Stem Cells and Early Mammalian Development. Cell Metab. 2018, 27, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreipke, R.; Wang, Y.; Miklas, J.W.; Mathieu, J.; Ruohola-baker, H. Metabolic Remodeling in early development and cardiomyocyte maturation. Semin. Cell Dev. Biol. 2016, 52, 84–92. [Google Scholar] [CrossRef]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.E.; Dai, D.F.; Laflamme, M.A. Human pluripotent stem cells: Prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv. Drug Deliv. Rev. 2016, 96, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinde, A.O.; Kantor, P.F.; Lopaschuk, G.D. Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Mol. Cell. Biochem. 1998, 188, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pohjoismäki, J.L.O.; Krüger, M.; Al-Furoukh, N.; Lagerstedt, A.; Karhunen, P.J.; Braun, T. Postnatal cardiomyocyte growth and mitochondrial reorganization cause multiple changes in the proteome of human cardiomyocytes. Mol. Biosyst. 2013, 9, 1210–1219. [Google Scholar] [CrossRef]
- Pereira, I.T.; Spangenberg, L.; Robert, A.W.; Amorín, R.; Stimamiglio, M.A.; Naya, H.; Dallagiovanna, B. Polysome profiling followed by RNA-seq of cardiac differentiation stages in hESCs. Sci. Data 2018, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pereira, I.T.; Spangenberg, L.; Robert, A.W.; Amorín, R.; Stimamiglio, M.A.; Naya, H.; Dallagiovanna, B. Cardiomyogenic differentiation is fine-tuned by differential mRNA association with polysomes. BMC Genom. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Robert, A.W.; Pereira, I.T.; Dallagiovanna, B.; Stimamiglio, M.A. Secretome Analysis Performed During in vitro Cardiac Differentiation: Discovering the Cardiac Microenvironment. Front. Cell Dev. Biol. 2020, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmer, B.M.; Eschenhagen, T. Human pluripotent stem cell-derived cardiomyocytes for studying energy metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 1. [Google Scholar] [CrossRef] [PubMed]
- Malandraki-Miller, S.; Lopez, C.A.; Al-Siddiqi, H.; Carr, C.A. Changing Metabolism in Differentiating Cardiac Progenitor Cells—Can Stem Cells Become Metabolically Flexible Cardiomyocytes? Front. Cardiovasc. Med. 2018, 5, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Wagner, R.A.; Wilson, K.D.; Xie, X.; Fu, J.; Drukker, M.; Lee, A.; Li, R.A.; Gambhir, S.S.; Weissman, I.L.; et al. Transcriptional and Functional Profiling of Human Embryonic Stem Cell-Derived Cardiomyocytes. PLoS ONE 2008, 3, e3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorzano, A.; Liesa, M.; Palacín, M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int. J. Biochem. Cell Biol. 2009, 41, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Anson, B.; Engle, S.; Will, Y. Characterization of human-induced pluripotent stem cell-derived cardiomyocytes: Bioenergetics and utilization in safety screening. Toxicol. Sci. 2012, 130, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef] [Green Version]
- Gilardi, F.; Desvergne, B. RXRs: Collegial partners. Subcell. Biochem. 2014. [Google Scholar] [CrossRef]
- Shulman, A.I.; Larson, C.; Mangelsdorf, D.J.; Ranganathan, R. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell 2004, 116, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Desvergne, B. RXR: From Partnership to Leadership in Metabolic Regulations. Vitam. Horm. 2007, 75, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Mangelsdorf, D.J. Liver X receptor signaling pathways in cardiovascular disease. Mol. Endocrinol. 2003, 17, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, P.A.; Kast, H.R.; Anisfeld, A.M. BAREing it all: The adoption of LXR and FXR and their roles in lipid homeostasis. J. Lipid Res. 2002, 43, 2–12. [Google Scholar]
- García, P.; Lorenzo, P.; De Lera, A.R. Natural Ligands of RXR Receptors, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 637, ISBN 9780128201442. [Google Scholar]
- Finck, B.N. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc. Res. 2007, 73, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.V.; van Gilst, W.H.; de Boer, R.A. Emerging role of liver X receptors in cardiac pathophysiology and heart failure. Basic Res. Cardiol. 2016, 111, 1–17. [Google Scholar] [CrossRef]
- Lin, Y.S.; Chang, T.H.; Shi, C.S.; Wang, Y.Z.; Ho, W.C.; Huang, H.-D.; Chang, S.T.; Pan, K.L.; Chen, M.C. Liver X receptor/retinoid X receptor pathway plays a regulatory role in pacing-induced cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e009146. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Yuan, A.; Shan, P.; Gao, E.; Wang, X.; Wang, Y.; Lau, W.B.; Koch, W.; Ma, X.L.; He, B. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur. Heart J. 2013, 34, 1834–1845. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Zhang, F.; Zhao, S.; Li, Y.; Chen, X.; Gao, E.; Xu, X.; Xiong, Z.; Zhang, X.; Zhang, J.; et al. Adiponectin determines farnesoid X receptor agonism-mediated cardioprotection against post-infarction remodelling and dysfunction. Cardiovasc. Res. 2018, 114, 1335–1349. [Google Scholar] [CrossRef]
- Czubryt, M.P.; Mcanally, J.; Fishman, G.I.; Olson, E.N. Regulation of peroxisome proliferator-activated receptor coactivator 1 (PGC-1) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2002, 100, 1711–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S. The function of vitamin D receptor in vitamin D action. J. Biochem. 2000, 127, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Hewison, M.; Gardner, D.G.; Wagner, C.L.; Sergeev, I.N.; Rutten, E.; Pittas, A.G.; Boland, R.; Ferrucci, L.; Bikle, D.D. Vitamin D: Beyond bone. Ann. N. Y. Acad. Sci. 2013, 1287, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Khammissa, R.A.G.; Fourie, J.; Motswaledi, M.H.; Ballyram, R.; Lemmer, J.; Feller, L. The Biological Activities of Vitamin D and Its Receptor in Relation to Calcium and Bone Homeostasis, Cancer, Immune and Cardiovascular Systems, Skin Biology, and Oral Health. BioMed Res. Int. 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.F.; Nikovits, W.; Bao, Z.Z.; Stockdale, F.E. Irx4 Forms an Inhibitory Complex with the Vitamin D and Retinoic X Receptors to Regulate Cardiac Chamber-specific slow MyHC3 Expression. J. Biol. Chem. 2001, 276, 28835–28841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 207–258. [Google Scholar] [CrossRef]
- Nakajima, Y. Retinoic acid signaling in heart development. Genesis 2019, 57, e23300. [Google Scholar] [CrossRef]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Metzler, M.A.; Sandell, L.L. Enzymatic metabolism of vitamin A in developing vertebrate embryos. Nutrients 2016, 8, 812. [Google Scholar] [CrossRef]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.I.; Cardoso, W.V.; Rosenthal, N.; Xavier-Neto, J. A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, J.B.; Xavier-Neto, J.; Shapiro, M.D.; Nayeem, S.M.; McCaffery, P.; Dräger, U.C.; Rosenthal, N. Dynamic patterns of retinoic acid synthesis and response in the developing mammalian heart. Dev. Biol. 1998, 199, 55–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.K.; Lee, S.A.; Belyaeva, O.V.; Wu, L.; Kedishvili, N.Y. Characterization of human short chain dehydrogenase/reductase SDR16C family members related to retinol dehydrogenase 10. Chem. Biol. Interact. 2017, 276, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Lynn, M.L.; Inman, K.E.; McDowell, W.; Trainor, P.A. RDH10 oxidation of vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS ONE 2012, 7, e30698. [Google Scholar] [CrossRef]
- Wu, L.; Belyaeva, O.V.; Adams, M.K.; Klyuyeva, A.V.; Lee, S.A.; Goggans, K.R.; Kesterson, R.A.; Popov, K.M.; Kedishvili, N.Y. Mice lacking the epidermal retinol dehydrogenases SDR16C5 and SDR16C6 display accelerated hair growth and enlarged meibomian glands. J. Biol. Chem. 2019, 294, 17060–17074. [Google Scholar] [CrossRef]
- Dupé, V.; Matt, N.; Garnier, J.M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C. Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes. J. Dev. Biol. 2020, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001, 15, 226–240. [Google Scholar] [CrossRef] [Green Version]
- Rydeen, A.B.; Waxman, J.S. Cyp26 enzymes are required to balance the cardiac and vascular lineages within the anterior lateral plate mesoderm. Dev. 2014, 141, 1638–1648. [Google Scholar] [CrossRef] [Green Version]
- Reijntjes, S.; Gale, E.; Maden, M. Expression of the retinoic acid catabolising enzyme CYP26B1 in the chick embryo and its regulation by retinoic acid. Gene Expr. Patterns 2003, 3, 621–627. [Google Scholar] [CrossRef]
- Reijntjes, S.; Gale, E.; Maden, M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev. Dyn. 2004, 230, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 2017, 173, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampron, C.; Rochette-Egly, C.; Gorry, P.; Dollé, P.; Mark, M.; Lufkin, T.; LeMeur, M.; Chambon, P. Mice deficient in cellular retinoic acid binding protein II (CRABPII) or in both CRABPI and CRABPII are essentially normal. Development 1995, 121, 539–548. [Google Scholar] [PubMed]
- Zolfaghari, R.; Ross, A.C. Lecithin:retinol acyltransferase from mouse and rat liver: cDNA cloning and liver-specific regulation by dietary vitamin A and retinoic acid. J. Lipid Res. 2000, 41, 2024–2034. [Google Scholar]
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, S.; Zaffran, S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev. 2017, 143, 9–19. [Google Scholar] [CrossRef]
- Polk, D.H. Thyroid hormone metabolism during development. Reprod. Fertil. Dev. 1995, 7, 469–477. [Google Scholar] [CrossRef]
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid Hormones and Cardiovascular Function and Diseases. J. Am. Coll. Cardiol. 2018, 71, 1781–1796. [Google Scholar] [CrossRef]
- Chattergoon, N.N. Thyroid hormone signaling and consequences for cardiac development. J. Endocrinol. 2019, 242, T145–T160. [Google Scholar] [CrossRef]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef] [Green Version]
- Dentice, M.; Morisco, C.; Vitale, M.; Rossi, G.; Fenzi, G.; Salvatore, D. The different cardiac expression of the type 2 iodothyronine deiodinase gene between human and rat is related to the differential response of the dio2 genes to Nkx-2.5 and GATA-4 transcription factors. Mol. Endocrinol. 2003, 17, 1508–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeöld, A.; Bianco, A.C. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, R.; Muller, A.; Simonides, W.S. Cardiac Thyroid Hormone Metabolism and Heart Failure. Eur. Thyroid J. 2017, 6, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Ng, K.M.; Chan, Y.C.; Lai, W.H.; Au, K.W.; Ho, C.Y.J.; Wong, L.Y.; Lau, C.P.; Tse, H.F.; Siu, C.W. Triiodothyronine promotes cardiac differentiation and maturation of embryonic stem cells via the classical genomic pathway. Mol. Endocrinol. 2010, 24, 1728–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, P.M. Physiological and molecular basis of Thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [Green Version]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Casas, F.; Pessemesse, L.; Grandemange, S.; Seyer, P.; Gueguen, N.; Baris, O.; Lepourry, L.; Cabello, G.; Wrutniak-Cabello, C. Overexpression of the Mitochondrial T3 Receptor p43 Induces a Shift in Skeletal Muscle Fiber Types. PLoS ONE 2008, 3, e2501. [Google Scholar] [CrossRef] [Green Version]
- Friesema, E.C.H.; Kuiper, G.G.J.M.; Jansen, J.; Visser, T.J.; Kester, M.H.A. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol. Endocrinol. 2006, 20, 2761–2772. [Google Scholar] [CrossRef] [Green Version]
- Cardim Pires, T.R.; Albanese, J.M.; Schwab, M.; Marette, A.; Carvalho, R.S.; Sola-Penna, M.; Zancan, P. Phosphofructokinase-P Modulates P44/42 MAPK Levels in HeLa Cells. J. Cell. Biochem. 2017, 118, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- André, E.; De Pauw, A.; Verdoy, R.; Brusa, D.; Bouzin, C.; Timmermans, A.; Bertrand, L.; Balligand, J.L. Changes of Metabolic Phenotype of Cardiac Progenitor Cells during Differentiation: Neutral Effect of Stimulation of AMP-Activated Protein Kinase. Stem Cells Dev. 2019, 28, 1498–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, R.W.; Patel, Y.M. Phosphoenolpyruvate Carboxykinase (GTP): The Gene and the Enzyme. In Advances in Enzymology and Related Areas of Molecular Biology; Wiley and Sons Inc.: Hoboken, NJ, USA, 1994; Volume 69, pp. 203–281. ISBN 9780470123157. [Google Scholar]
- Hakimi, P.; Yang, J.; Casadesus, G.; Massillon, D.; Tolentino-Silva, F.; Nye, C.K.; Cabrera, M.E.; Hagen, D.R.; Utter, C.B.; Baghdy, Y.; et al. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J. Biol. Chem. 2007, 282, 32844–32855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakugawa, S.; Langton, P.F.; Zebisch, M.; Howell, S.A.; Chang, T.H.; Liu, Y.; Feizi, T.; Bineva, G.; O’Reilly, N.; Snijders, A.P.; et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature 2015, 519, 187–192. [Google Scholar] [CrossRef]
- Gessert, S.; Kühl, M. The multiple phases and faces of Wnt signaling during cardiac differentiation and development. Circ. Res. 2010, 107, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.M.J.; Hession, C.; Johansen, B.; Hayes, G.; McGray, P.; Chow, E.P.; Tizard, R.; Pepinsky, R.B. Structure and properties of a human non-pancreatic phospholipase A2. J. Biol. Chem. 1989, 264, 5768−5775. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Sato, H.; Yamamoto, K. Secreted phospholipase A 2 revisited. J. Biochem. 2011, 150, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Zhu, K.; Fujita, C.K.; Zhao, M.; Lam, K.S.; Kurth, M.J.; Takada, Y.K.; Takada, Y. Proinflammatory secreted phospholipase A2 type IIA (sPLA-IIA) induces integrin activation through direct binding to a newly identified binding site (site 2) in integrins αvβ3, α4β1, and α5β1. J. Biol. Chem. 2015, 290, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Snitko, Y.; Yoon, E.T.; Cho, W. High specificity of human secretory class II phospholipase A2 for phosphatidic acid. Biochem. J. 1997, 321, 737–741. [Google Scholar] [CrossRef] [Green Version]
- Divchev, D.; Schieffer, B. The secretory phospholipase A2 group IIA: A missing link between inflammation, activated renin-angiotensin system, and atherogenesis? Vasc. Health Risk Manag. 2008, 4, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Kuefner, M.S.; Pham, K.; Redd, J.R.; Stephenson, E.J.; Harvey, I.; Deng, X.; Bridges, D.; Boilard, E.; Elam, M.B.; Park, E.A. Secretory phospholipase A2 group IIA modulates insulin sensitivity and metabolism. J. Lipid Res. 2017, 58, 1822–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanguy, E.; Wang, Q.; Moine, H.; Vitale, N. Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front. Cell. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickard, R.T.; Strifler, B.A.; Kramer, R.M.; Sharp, J.D. Molecular cloning of two new human paralogs of 85-kDa cytosolic phospholipase A2. J. Biol. Chem. 1999, 274, 8823–8831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underwood, K.W.; Song, C.; Kriz, R.W.; Chang, X.J.; Knopf, J.L.; Lin, L.L. A novel calcium-independent phospholipase A2, cPLA2-γ, that is prenylated and contains homology to cPLA2. J. Biol. Chem. 1998, 273, 21926–21932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, A.; Ghosh, M.; Spencer, D.M.; Leslie, C.C. Enzymatic properties of human cytosolic phospholipase A2γ. J. Biol. Chem. 2002, 277, 29526–29536. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Kamata, R.; Kawagishi, N.; Nakanishi, H.; Suzuki, H.; Sugiura, T. Roles of C-Terminal Processing, and Involvement in Transacylation Reaction of Human Group IVC Phospholipase A2 (cPLA2γ). J. Biochem. 2005, 137, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Tanaka, K.; Kamata, R.; Kumazawa, T.; Suzuki, N.; Koga, H.; Waku, K.; Sugiura, T. Subcellular localization and lysophospholipase/transacylation activities of human group IVC phospholipase A2 (cPLA2gamma). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 1011–1022. [Google Scholar] [CrossRef]
- Ammar, M.R.; Kassas, N.; Bader, M.F.; Vitale, N. Phosphatidic acid in neuronal development: A node for membrane and cytoskeleton rearrangements. Biochimie 2014, 107, 51–57. [Google Scholar] [CrossRef]
- Pascual, F.; Carman, G.M. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2013, 1831, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. 2017, 161, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Gellatly, S.A.; Kalujnaia, S.; Cramb, G. Cloning, tissue distribution and sub-cellular localisation of phospholipase C X-domain containing protein (PLCXD) isoforms. Biochem. Biophys. Res. Commun. 2012, 424, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Traer, E.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. The cloning and characterization of a novel human diacylglycerol kinase, DGKι. J. Biol. Chem. 1998, 273, 32746–32752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu-Sekine, B.; Raben, D.M. Regulation and roles of neuronal diacylglycerol kinases: A lipid perspective. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Kagaya, Y.; Takahashi, J.; Sugie, T.; Ohta, J.; Watanabe, J.; Shirato, K.; Kondo, H.; Goto, K. Gene expression and in situ localization of diacylglycerol kinase isozymes in normal and infarcted rat hearts effects of captopril treatment. Circ. Res. 2001, 89, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 325, pp. 215–273. ISBN 9780128048061. [Google Scholar]
- Kreuger, J.; Kjellén, L. Heparan Sulfate Biosynthesis: Regulation and Variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Kraushaar, D.C.; Dalton, S.; Wang, L. Heparan sulfate: A key regulator of embryonic stem cell fate. Biol. Chem. 2013, 394, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.N.; Lombaert, I.M.A.; Cowherd, S.N.; Shworak, N.W.; Xu, Y.; Liu, J.; Hoffman, M.P. Hs3st3-Modified Heparan Sulfate Controls KIT+ Progenitor Expansion by Regulating 3-O-Sulfotransferases. Dev. Cell 2014, 29, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Sasse, P.; Malan, D.; Fleischmann, M.; Roell, W.; Gustafsson, E.; Bostani, T.; Fan, Y.; Kolbe, T.; Breitbach, M.; Addicks, K.; et al. Perlecan is critical for heart stability. Cardiovasc. Res. 2008, 80, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, Y.; Nakada, S.; Yoshihara, T.; Nara, T.; Furuya, N.; Miida, T.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan, a heparan sulfate proteoglycan, regulates systemic metabolism with dynamic changes in adipose tissue and skeletal muscle. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- McDonnell, K.M.W.; Grow, W.A. Reduced glycosaminoglycan sulfation diminishes the agrin signal transduction pathway. Dev. Neurosci. 2004, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Le Bouter, S.; Szuts, V.; Varro, A.; Escande, D.; Nattel, S.; Demolombe, S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. 2007, 582, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Tharp, D.L.; Georger, M.A.; Slivano, O.J.; Lee, M.Y.; Wamhoff, B.R.; Bowles, D.K.; Miano, J.M. The smooth muscle cell-restricted KCNMB1 ion channel subunit is a direct transcriptional target of serum response factor and myocardin. J. Biol. Chem. 2009, 284, 33671–33682. [Google Scholar] [CrossRef] [Green Version]
- Dib-Hajj, S.D.; Yang, Y.; Black, J.A.; Waxman, S.G. The Na v 1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 2013, 14, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Markunas, C.A.; Quinn, K.S.; Collins, A.L.; Garrett, M.E.; Lachiewicz, A.M.; Sommer, J.L.; Morrissey-Kane, E.; Kollins, S.H.; Anastopoulos, A.D.; Ashley-Koch, A.E. Genetic variants in SLC9A9 are associated with measures of attention-deficit/hyperactivity disorder symptoms in families. Psychiatr. Genet. 2010, 20, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taegtmeyer, H. Glycogen in the heart-An expanded view. J. Mol. Cell. Cardiol. 2004, 37, 7–10. [Google Scholar] [CrossRef]
- Pederson, B.A.; Chen, H.; Schroeder, J.M.; Shou, W.; DePaoli-Roach, A.A.; Roach, P.J. Abnormal Cardiac Development in the Absence of Heart Glycogen. Mol. Cell. Biol. 2004, 24, 7179–7187. [Google Scholar] [CrossRef] [Green Version]
- Crosson, S.M.; Khan, A.; Printen, J.; Pessin, J.E.; Saltiel, A.R. PTG gene deletion causes impaired glycogen synthesis and. Digestion 2003, 111, 1423–1432. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Energy Metabolism of the Heart: From Basic Concepts To Clinical. Curr. Probl. Cardiol. 1994, 19, 61–86. [Google Scholar] [CrossRef]
- Smith, F.M.; Garfield, A.S.; Ward, A. Regulation of growth and metabolism by imprinted genes. Cytogenet. Genome Res. 2006, 113, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.P.; Hughes, J.; Hassan, A.B.; Brüning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells 2019, 8, 1207. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, T.; Hollander, J.M.; Nakamura, D.S.; Liu, Z.; Rosen, C.J.; Georgakoudi, I.; Zeng, L. An essential role for IGF2 in cartilage development and glucose metabolism during postnatal long bone growth. Dev. 2017, 144, 3533–3546. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Toole, B.P.; Dealy, C.N.; Kosher, R.A. Hyaluronan in limb morphogenesis. Dev. Biol. 2007, 305, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.K.; Rolle, M.W.; Potter-Perigo, S.; Braun, K.R.; Van Biber, B.P.; Laflamme, M.A.; Murry, C.E.; Wight, T.N. Differentiation of cardiomyocytes from human embryonic stem cells is accompanied by changes in the extracellular matrix production of versican and hyaluronan. J. Cell. Biochem. 2010, 111, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.M.M.; Chen, A.; Koh, P.W.W.; Deng, T.Z.Z.; Sinha, R.; Tsai, J.M.M.; Barkal, A.A.A.; Shen, K.Y.Y.; Jain, R.; Morganti, R.M.M.; et al. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell 2016, 166, 451–468. [Google Scholar] [CrossRef] [Green Version]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Castan-Laurell, I.; Masri, B.; Valet, P. The apelin/APJ system as a therapeutic target in metabolic diseases. Expert Opin. Ther. Targets 2019, 23, 215–225. [Google Scholar] [CrossRef]
- Kuba, K.; Sato, T.; Imai, Y.; Yamaguchi, T. Apelin and Elabela/Toddler; double ligands for APJ/Apelin receptor in heart development, physiology, and pathology. Peptides 2019, 111, 62–70. [Google Scholar] [CrossRef]
- Scott, I.C.; Masri, B.; D’Amico, L.A.; Jin, S.W.; Jungblut, B.; Wehman, A.M.; Baier, H.; Audigier, Y.; Stainier, D.Y.R. The G Protein-Coupled Receptor Agtrl1b Regulates Early Development of Myocardial Progenitors. Dev. Cell 2007, 12, 403–413. [Google Scholar] [CrossRef]
- Paskaradevan, S.; Scott, I.C. The Aplnr GPCR regulates myocardial progenitor development via a novel cell-non-autonomous, G i/o protein-independent pathway. Biol. Open 2012, 1, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Christalla, P.; Hudson, J.E.; Zimmermann, W.-H. The Cardiogenic Niche as a Fundamental Building Block of Engineered Myocardium. Cells Tissues Organs 2012, 195, 82–93. [Google Scholar] [CrossRef]
- Leitolis, A.; Robert, A.W.; Pereira, I.T.; Correa, A.; Stimamiglio, M.A.; Carr, C.; Robert, A.W. Cardiomyogenesis Modeling Using Pluripotent Stem Cells: The Role of Microenvironmental Signaling. Front. Cell Dev. Biol. 2019, 7, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Chung, K.W.; Jeong, H.O.; Lee, B.; Hyun, D.; Park, J.W.; Kim, S.M.; Yu, B.P.; Chung, H.Y. MMP2-A2M interaction increases ECM accumulation in aged rat kidney and its modulation by calorie restriction. Oncotarget 2018, 9, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Han, P.F.; Che, X.-D.; Li, H.Z.; Gao, Y.Y.; Wei, X.C.; Li, P.C. Annexin A1 involved in the regulation of inflammation and cell signaling pathways. Chin. J. Traumatol. 2020, 23, 96–101. [Google Scholar] [CrossRef]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the many talents of an old protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef] [Green Version]
- Bizzarro, V.; Belvedere, R.; Migliaro, V.; Romano, E.; Parente, L.; Petrella, A. Hypoxia regulates ANXA1 expression to support prostate cancer cell invasion and aggressiveness. Cell Adhes. Migr. 2017, 11, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Elliott, D.A.; Braam, S.R.; Koutsis, K.; Ng, E.S.; Jenny, R.; Lagerqvist, E.L.; Biben, C.; Hatzistavrou, T.; Hirst, C.E.; Yu, Q.C.; et al. NKX2-5eGFP/w hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods 2011, 8, 1037–1040. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barisón, M.J.; Pereira, I.T.; Waloski Robert, A.; Dallagiovanna, B. Reorganization of Metabolism during Cardiomyogenesis Implies Time-Specific Signaling Pathway Regulation. Int. J. Mol. Sci. 2021, 22, 1330. https://doi.org/10.3390/ijms22031330
Barisón MJ, Pereira IT, Waloski Robert A, Dallagiovanna B. Reorganization of Metabolism during Cardiomyogenesis Implies Time-Specific Signaling Pathway Regulation. International Journal of Molecular Sciences. 2021; 22(3):1330. https://doi.org/10.3390/ijms22031330
Chicago/Turabian StyleBarisón, María Julia, Isabela Tiemy Pereira, Anny Waloski Robert, and Bruno Dallagiovanna. 2021. "Reorganization of Metabolism during Cardiomyogenesis Implies Time-Specific Signaling Pathway Regulation" International Journal of Molecular Sciences 22, no. 3: 1330. https://doi.org/10.3390/ijms22031330
APA StyleBarisón, M. J., Pereira, I. T., Waloski Robert, A., & Dallagiovanna, B. (2021). Reorganization of Metabolism during Cardiomyogenesis Implies Time-Specific Signaling Pathway Regulation. International Journal of Molecular Sciences, 22(3), 1330. https://doi.org/10.3390/ijms22031330