
Spermidine Attenuates Oxidative Stress-Induced Apoptosis via Blocking Ca2+ Overload in Retinal Pigment Epithelial Cells Independently of ROS


 ,
,  ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
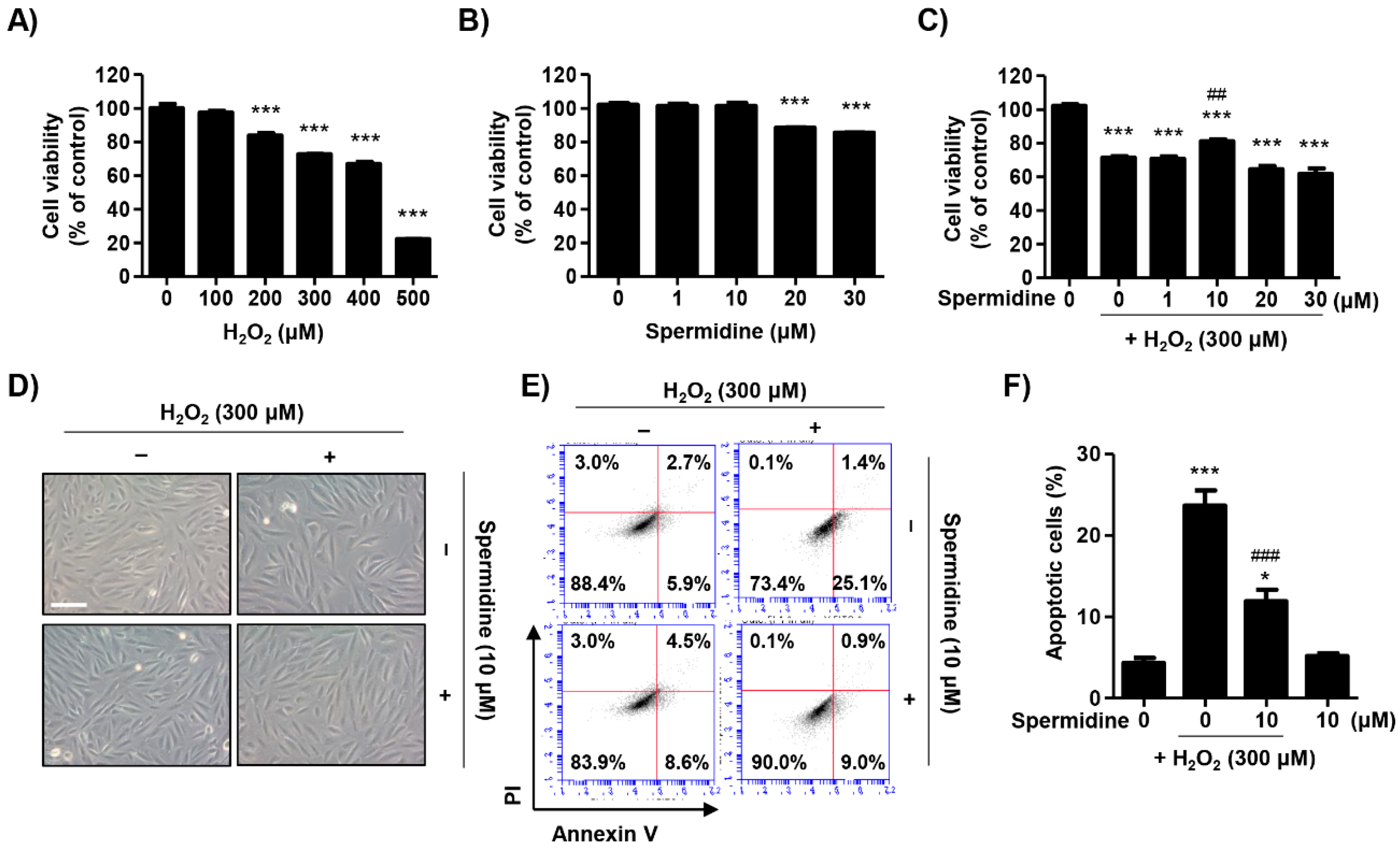
2.1. Spermidine Attenuated H2O2-Induced Cytotoxicity in ARPE-19 Cells
2.2. Spermidine Downregulated Extrinsic and Intrinsic Apoptosis Pathways in H2O2-Stimulated ARPE-19 Cells
2.3. Spermidine Suppressed DNA Damage and Dysregulation of Cell Cycle Processes in H2O2-Stimulated ARPE-19 Cells
2.4. Spermidine Had Antioxidant Capacity But Did Not Regulate Intracellular ROS Generation in H2O2-Stimulated ARPE-19 Cells
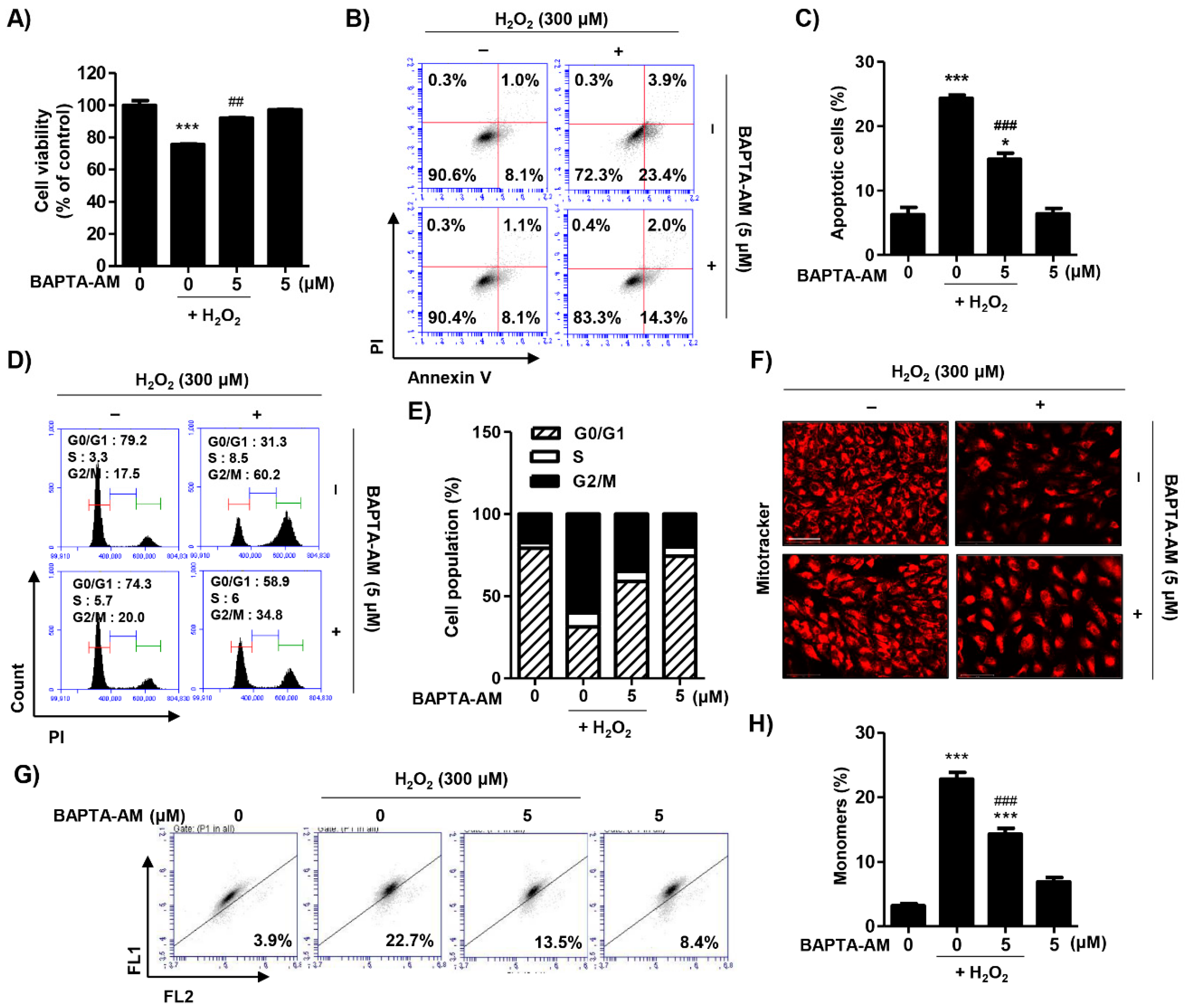
2.5. Spermidine Decreased Cytosolic Ca2+ Levels Released Due to Endoplasmic Reticulum (ER) Stress in H2O2-Stimulated ARPE-19 Cells
2.6. Blocking of Cytosolic Ca2+ Levels Attenuated H2O2-Induced Cytotoxicity in ARPE-19 Cells
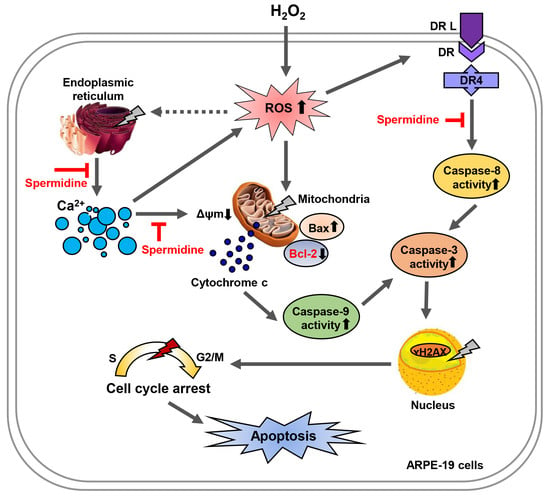
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell culture and Spermidine Treatment
4.3. Cell Viability Analysis
4.4. Flow Cytometric Analysis
4.5. Intracellular ROS Detection
4.6. Fluorescence Image Analysis
4.7. Western Blot Analysis
4.8. Measurement of Caspases Activities
4.9. Immunofluorescence Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845. [Google Scholar] [CrossRef]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef]
- Stahl, A. The diagnosis and treatment of age-related macular degeneration. Dtsch. Arztebl. Int. 2020, 117, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef]
- Du, W.; An, Y.; He, X.; Zhang, D.; He, W. Protection of kaempferol on oxidative stress-induced retinal pigment epithelial cell damage. Oxid. Med. Cell. Longev. 2018, 2018, 1610751. [Google Scholar] [CrossRef]
- Al-Zamil, W.M.; Yassin, S.A. Recent developments in age-related macular degeneration: A review. Clin. Interv. Aging 2017, 12, 1313–1330. [Google Scholar] [CrossRef]
- Liu, Y.; Li, R.; Xie, J.; Hu, J.; Huang, X.; Ren, F.; Li, L. Protective effect of hydrogen on sodium iodate-induced age-related macular degeneration in mice. Front. Aging Neurosci. 2018, 10, 389. [Google Scholar] [CrossRef]
- Dieguez, H.H.; Romeo, H.E.; Alaimo, A.; González Fleitas, M.F.; Aranda, M.L.; Rosenstein, R.E.; Dorfman, D. Oxidative stress damage circumscribed to the central temporal retinal pigment epithelium in early experimental non-exudative age-related macular degeneration. Free Radic. Biol. Med. 2019, 131, 72–80. [Google Scholar] [CrossRef]
- Patocka, J.; Kuehn, G.D. Natural polyamines and their biological consequence in mammals. Acta Med. 2000, 43, 119–124. [Google Scholar] [CrossRef]
- Perez-Leal, O.; Merali, S. Regulation of polyamine metabolism by translational control. Amino Acids 2012, 42, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, N.; Moriwaki, M.; Shiraki, K.; Miki, T.; Otani, S. The involvement of polyamines in the proliferation of cultured retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1975–1983. [Google Scholar]
- Johnson, D.A.; Fields, C.; Fallon, A.; Fitzgerald, M.E.; Viar, M.J.; Johnson, L.R. Polyamine-dependent migration of retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1228–1233. [Google Scholar]
- Laube, G.; Veh, R.W. Astrocytes, not neurons, show most prominent staining for spermidine/spermine-like immunoreactivity in adult rat brain. Glia 1997, 19, 171–179. [Google Scholar] [CrossRef]
- Biedermann, B.; Skatchkov, S.N.; Brunk, I.; Bringmann, A.; Pannicke, T.; Bernstein, H.G.; Faude, F.; Germer, A.; Veh, R.; Reichenbach, A. Spermine/spermidine is expressed by retinal glial (Müller) cells, and controls distinct K+ channels of their membrane. Glia 1998, 23, 209–220. [Google Scholar] [CrossRef]
- Noro, T.; Namekata, K.; Kimura, A.; Guo, X.; Azuchi, Y.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis. 2015, 6, e1720. [Google Scholar]
- Noro, T.; Namekata, K.; Azuchi, Y.; Kimura, A.; Guo, X.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine ameliorates neurodegeneration in a mouse model of normal tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5012–5019. [Google Scholar] [CrossRef]
- Rider, J.E.; Hacker, A.; Mackintosh, C.A.; Pegg, A.E.; Woster, P.M.; Casero, R.A., Jr. Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids 2007, 33, 231–240. [Google Scholar] [CrossRef]
- Guo, X.; Harada, C.; Namekata, K.; Kimura, A.; Mitamura, Y.; Yoshida, H.; Matsumoto, Y.; Harada, T. Spermidine alleviates severity of murine experimental autoimmune encephalomyelitis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2696–2703. [Google Scholar] [CrossRef]
- Dunn, K.C.; Aotaki-Keen, A.E.; Putkey, F.R.; Hjelmeland, L.M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 1996, 62, 155–169. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Rózanowska, M.; Jarvis-Evans, J.; Korytowski, W.; Boulton, M.E.; Burke, J.M.; Sarna, T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem. 1995, 270, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Tate, D.J., Jr.; Miceli, M.V.; Newsome, D.A. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1271–1279. [Google Scholar]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008, 480, 85–94. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, R.; Ye, M.; Zhang, L. Genipin protects against H2O2-induced oxidative damage in retinal pigment epithelial cells by promoting Nrf2 signaling. Int. J. Mol. Med. 2019, 43, 936–944. [Google Scholar]
- Cui, R.; Tian, L.; Lu, D.; Li, H.; Cui, J. Exendin-4 protects human retinal pigment epithelial cells from H2O2-induced oxidative damage via activation of NRF2 signaling. Ophthalmic Res. 2020, 63, 404–412. [Google Scholar] [CrossRef]
- Weigel, A.L.; Handa, J.T.; Hjelmeland, L.M. Microarray analysis of H2O2-, HNE-, or tBH-treated ARPE-19 cells. Free Radic. Biol. Med. 2002, 33, 1419–1432. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Kalinowska-Hero, K.M.; Widlak, P. The major apoptotic endonuclease DFF40/CAD is a deoxyribose-specific and double-strand-specific enzyme. Apoptosis 2008, 13, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef]
- Nashine, S.; Kenney, M.C. Effects of mitochondrial-derived peptides (MDPs) on mitochondrial and cellular health in AMD. Cells 2020, 9, 1102. [Google Scholar] [CrossRef] [PubMed]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [PubMed]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- Pearce, A.K.; Humphrey, T.C. Integrating stress-response and cell-cycle checkpoint pathways. Trends Cell Biol. 2001, 11, 426–433. [Google Scholar] [PubMed]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair 2004, 3, 959–967. [Google Scholar] [CrossRef]
- Stark, G.R.; Taylor, W.R. Control of the G2/M transition. Mol. Biotechnol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Jorgensen, P.; Tyers, M. How cells coordinate growth and division. Curr. Biol. 2004, 14, R1014–R1027. [Google Scholar] [CrossRef]
- Li, L.; Gu, B.; Zhou, F.; Chi, J.; Wang, F.; Peng, G.; Xie, F.; Qing, J.; Feng, D.; Lu, S.; et al. Human herpesvirus 6 suppresses T cell proliferation through induction of cell cycle arrest in infected cells in the G2/M phase. J. Virol. 2011, 85, 6774–6783. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, W.; Zhou, X.; Long, C.; Kuang, X.; Hu, J.; Tang, Y.; Liu, L.; He, J.; Huang, Z.; et al. Protective effect of lutein on ARPE-19 cells upon H2O2-induced G2/M arrest. Mol. Med. Rep. 2017, 16, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, K.K.; Tong, Y.; Zhou, Y.L.; Wang, Y.X.; Zhao, P.Q.; Wang, Z.Y. Exogenous NAD(+) decreases oxidative stress and protects H2O2-treated RPE cells against necrotic death through the up-regulation of autophagy. Sci. Rep. 2016, 6, 26322. [Google Scholar]
- Park, C.; Lee, H.; Hong, S.H.; Kim, J.H.; Park, S.K.; Jeong, J.W.; Kim, G.Y.; Hyun, J.W.; Yun, S.J.; Kim, B.W.; et al. Protective effect of diphlorethohydroxycarmalol against oxidative stress-induced DNA damage and apoptosis in retinal pigment epithelial cells. Cutan. Ocul. Toxicol. 2019, 38, 298–308. [Google Scholar] [CrossRef]
- Droin, N.; Dubrez, L.; Eymin, B.; Renvoizé, C.; Bréard, J.; Dimanche-Boitrel, M.T.; Solary, E. Upregulation of CASP genes in human tumor cells undergoing etoposide-induced apoptosis. Oncogene 1998, 16, 2885–2894. [Google Scholar] [CrossRef]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Cano, M.; Wang, L.; Wan, J.; Barnett, B.P.; Ebrahimi, K.; Qian, J.; Handa, J.T. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Radic. Biol. Med. 2014, 69, 1–14. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Hyttinen, J.M.; Toropainen, E.; Kaarniranta, K. Endoplasmic reticulum stress in age-related macular degeneration: Trigger for neovascularization. Mol. Med. 2010, 16, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Bi, H.E.; Sheng, Y.; Cheng, L.B.; Wendu, R.L.; Wang, C.H.; Cao, G.F.; Jiang, Q. Ultraviolet (UV) and hydrogen peroxide activate ceramide-ER stress-AMPK signaling axis to promote retinal pigment epithelium (RPE) cell apoptosis. Int. J. Mol. Sci. 2013, 14, 10355–10368. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Fan, B.; Zheng, Y.C. Calcium overload is a critical step in programmed necrosis of ARPE-19 cells induced by high-concentration H2O2. Biomed. Environ. Sci. 2010, 23, 371–377. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Gong, L. Gamma linolenic acid suppresses hypoxia-induced proliferation and invasion of non-small cell lung cancer cells by inhibition of HIF1α. Genes Genom. 2020, 42, 927–935. [Google Scholar]
- Zhang, N.; Li, F.; Gao, J.; Zhang, S.; Wang, Q. Osteopontin accelerates the development and metastasis of bladder cancer via activating JAK1/STAT1 pathway. Genes Genom. 2020, 42, 467–475. [Google Scholar] [CrossRef]
- Hwangbo, H.; Kim, S.Y.; Lee, H.; Park, S.H.; Hong, S.H.; Park, C.; Kim, G.Y.; Leem, S.H.; Hyun, J.W.; Cheong, J.; et al. Auranofin enhances sulforaphane-mediated apoptosis in hepatocellular carcinoma Hep3B cells through inactivation of the PI3K/Akt signaling pathway. Biomol. Ther. 2020, 28, 443–455. [Google Scholar] [CrossRef]
- Hong, S.H.; Cha, H.J.; Hwang-Bo, H.; Kim, M.Y.; Kim, S.Y.; Ji, S.Y.; Cheong, J.; Park, C.; Lee, H.; Kim, G.Y.; et al. Anti-proliferative and pro-apoptotic effects of licochalcone A through ROS-mediated cell cycle arrest and apoptosis in human bladder cancer cells. Int. J. Mol. Sci. 2019, 20, 3820. [Google Scholar] [CrossRef]
- Hou, N.; He, X.; Yang, Y.; Fu, J.; Zhang, W.; Guo, Z.; Hu, Y.; Liang, L.; Xie, W.; Xiong, H.; et al. TRPV1 induced apoptosis of colorectal cancer cells by activating calcineurin-NFAT2-p53 signaling pathway. Biomed. Res. Int. 2019, 2019, 6712536. [Google Scholar]
- Noh, Y.; Ahn, J.H.; Lee, J.W.; Hong, J.; Lee, T.K.; Kim, B.; Kim, S.S.; Won, M.H. Brain Factor-7® improves learning and memory deficits and attenuates ischemic brain damage by reduction of ROS generation in stroke in vivo and in vitro. Lab. Anim. Res. 2020, 36, 24. [Google Scholar] [CrossRef]
- Park, S.; Kim, M.; Hong, Y.; Lee, H.; Tran, Q.; Kim, C.; Kwon, S.H.; Park, J.; Park, J.; Kim, S.H. Myristoylated TMEM39AS41, a cell-permeable peptide, causes lung cancer cell death. Toxicol. Res. 2020, 36, 123–130. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.H.; Kim, J.-H.; Hwangbo, H.; Kim, S.Y.; Ji, S.Y.; Kim, M.Y.; Cha, H.-J.; Park, C.; Hong, S.H.; Kim, G.-Y.; et al. Spermidine Attenuates Oxidative Stress-Induced Apoptosis via Blocking Ca2+ Overload in Retinal Pigment Epithelial Cells Independently of ROS. Int. J. Mol. Sci. 2021, 22, 1361. https://doi.org/10.3390/ijms22031361
Kim DH, Kim J-H, Hwangbo H, Kim SY, Ji SY, Kim MY, Cha H-J, Park C, Hong SH, Kim G-Y, et al. Spermidine Attenuates Oxidative Stress-Induced Apoptosis via Blocking Ca2+ Overload in Retinal Pigment Epithelial Cells Independently of ROS. International Journal of Molecular Sciences. 2021; 22(3):1361. https://doi.org/10.3390/ijms22031361
Chicago/Turabian StyleKim, Da Hye, Jeong-Hwan Kim, Hyun Hwangbo, So Young Kim, Seon Yeong Ji, Min Yeong Kim, Hee-Jae Cha, Cheol Park, Su Hyun Hong, Gi-Young Kim, and et al. 2021. "Spermidine Attenuates Oxidative Stress-Induced Apoptosis via Blocking Ca2+ Overload in Retinal Pigment Epithelial Cells Independently of ROS" International Journal of Molecular Sciences 22, no. 3: 1361. https://doi.org/10.3390/ijms22031361
APA StyleKim, D. H., Kim, J.-H., Hwangbo, H., Kim, S. Y., Ji, S. Y., Kim, M. Y., Cha, H.-J., Park, C., Hong, S. H., Kim, G.-Y., Park, S.-K., Jeong, J.-W., Kim, M.-Y., Choi, Y. H., & Lee, H. (2021). Spermidine Attenuates Oxidative Stress-Induced Apoptosis via Blocking Ca2+ Overload in Retinal Pigment Epithelial Cells Independently of ROS. International Journal of Molecular Sciences, 22(3), 1361. https://doi.org/10.3390/ijms22031361