
Cancer-Targeted Controlled Delivery of Chemotherapeutic Anthracycline Derivatives Using Apoferritin Nanocage Carriers


Abstract
:
1. Introduction
2. Results
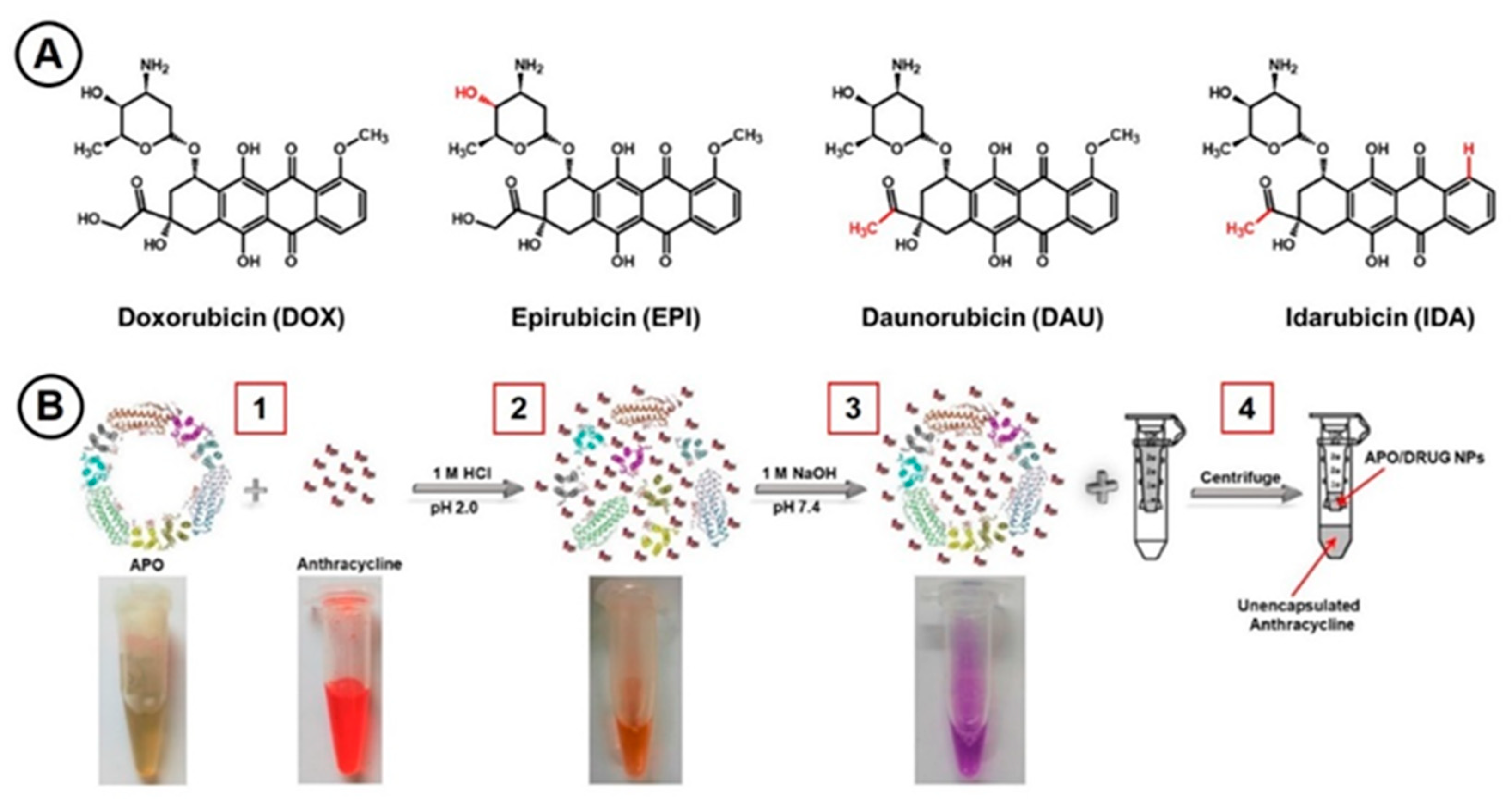
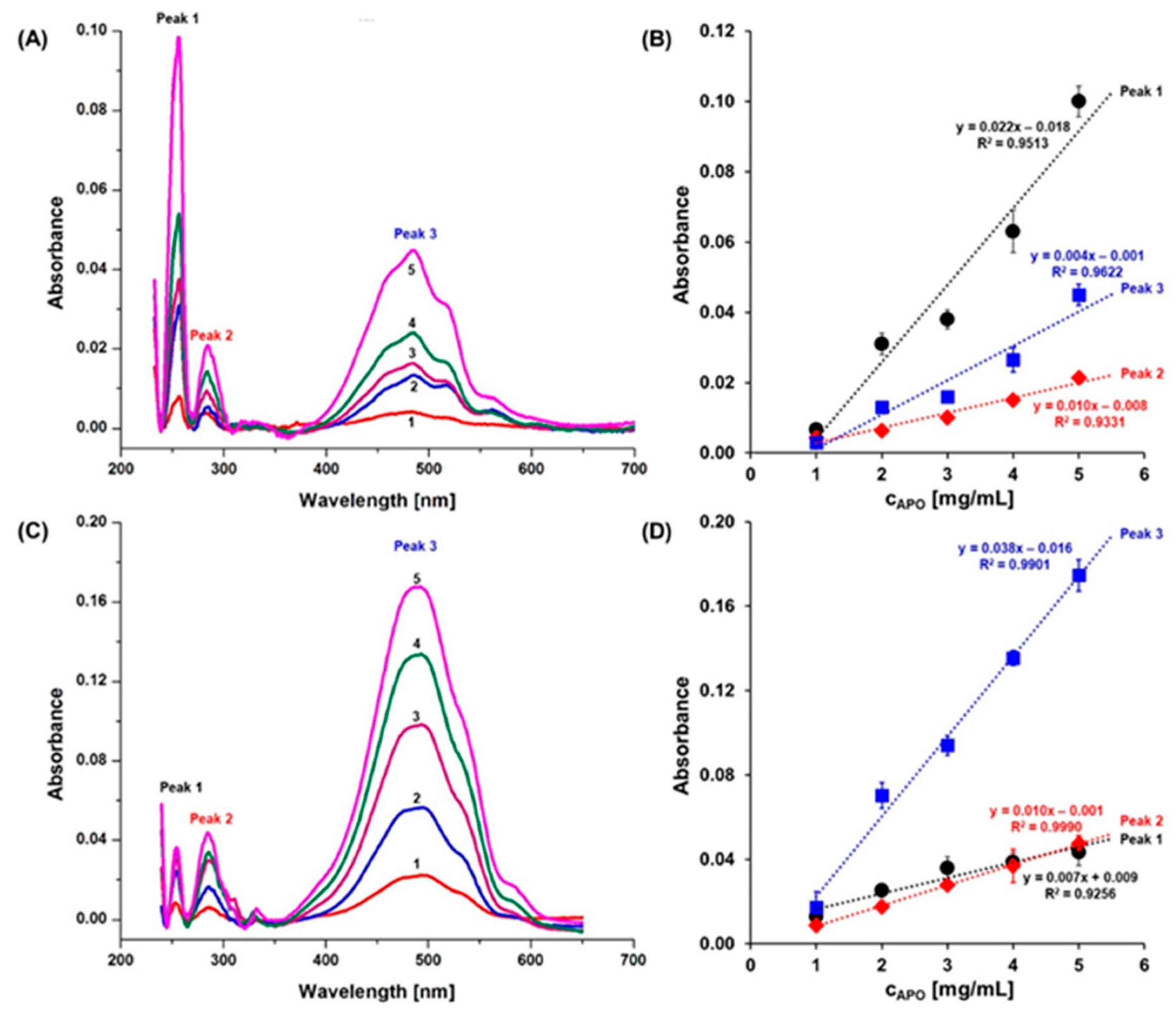
2.1. Anthracycline Encapsulation in Apoferritin Nanocages
2.2. Circular Dichroism Spectroscopy
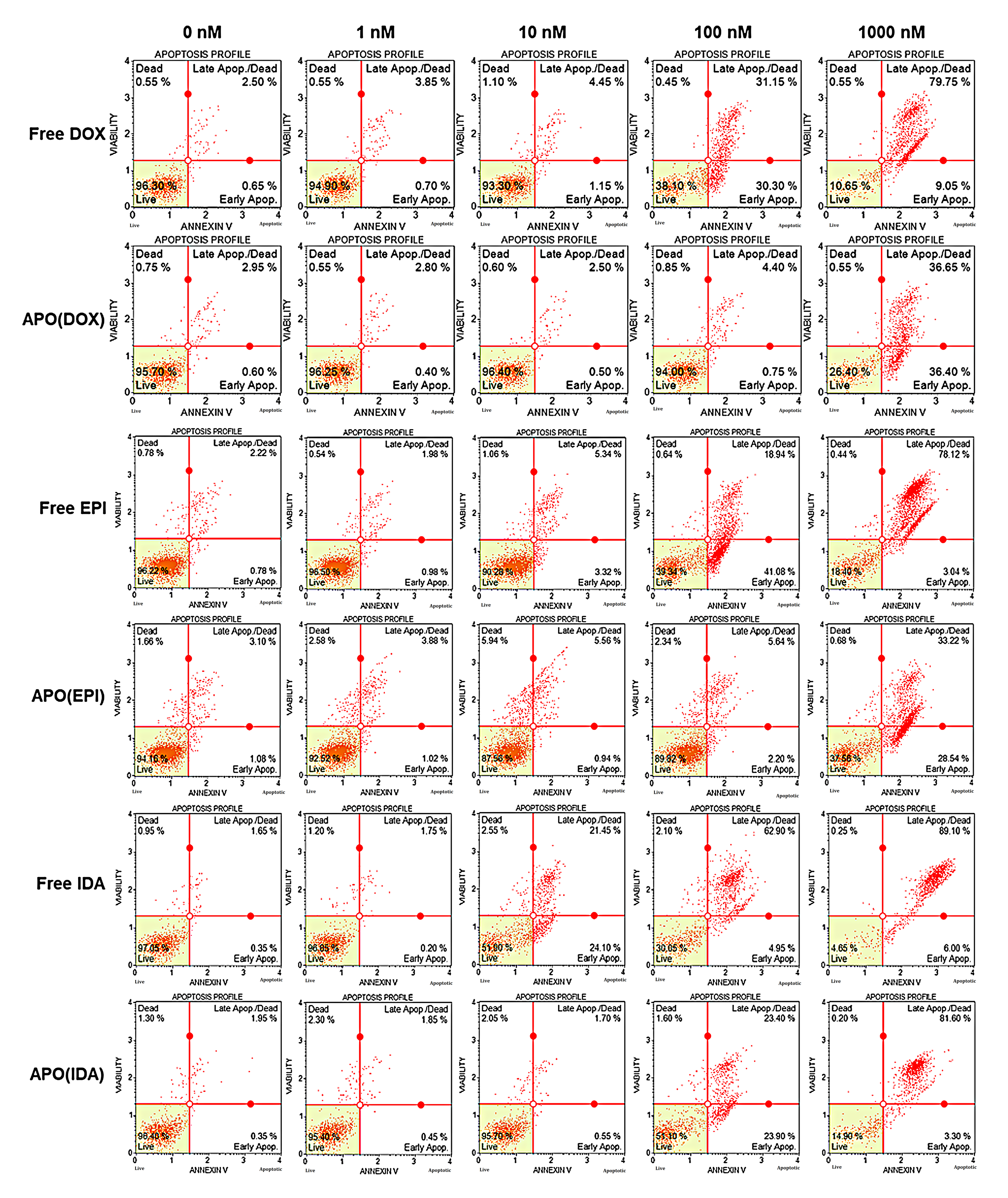
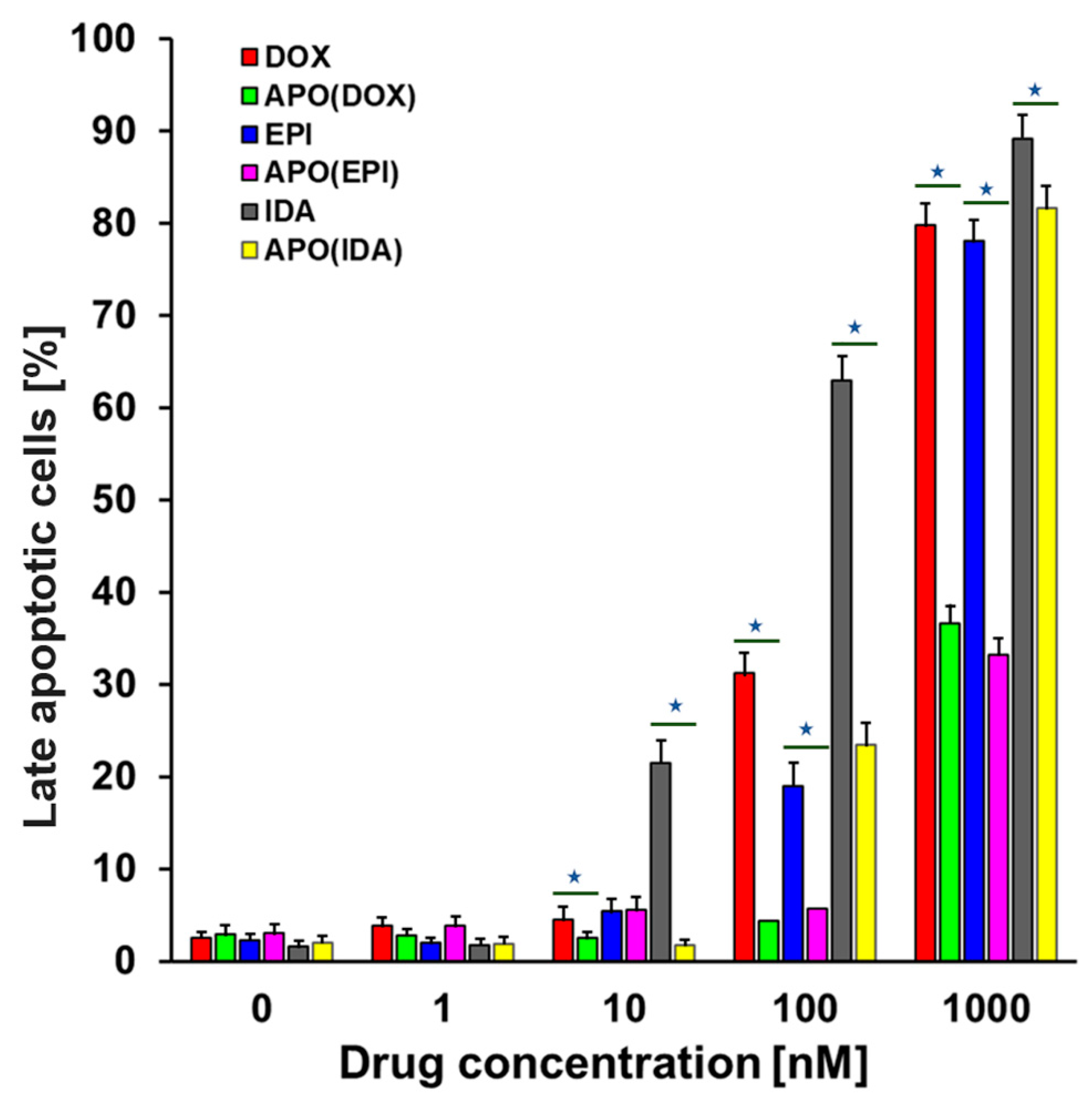
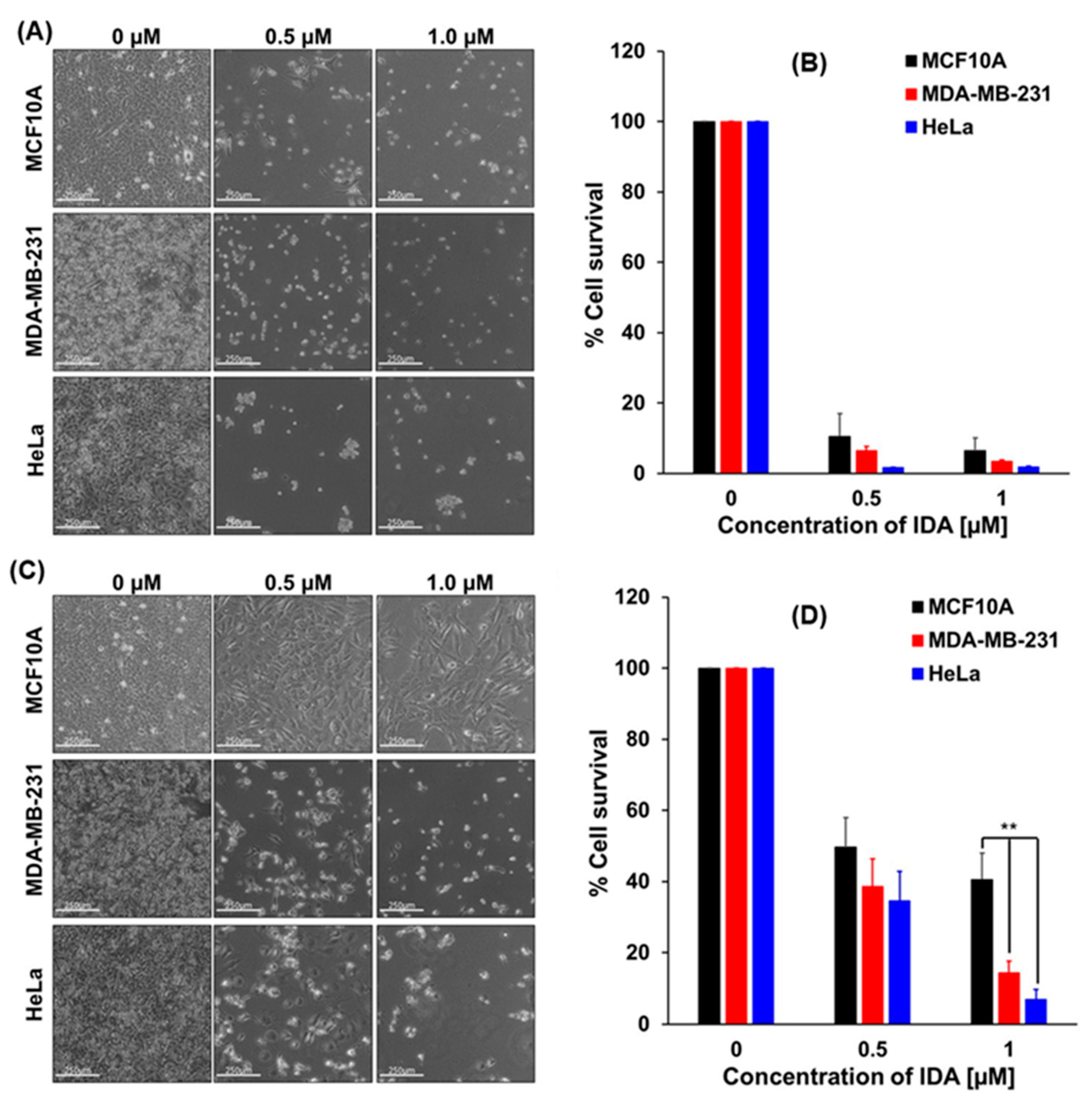
2.3. Cell Death Assay
2.4. Cytotoxicity/Growth Inhibition Assay by Crystal Violet Staining
2.5. Determination of the Dose–Response Relationships and EC50 Values
3. Discussion
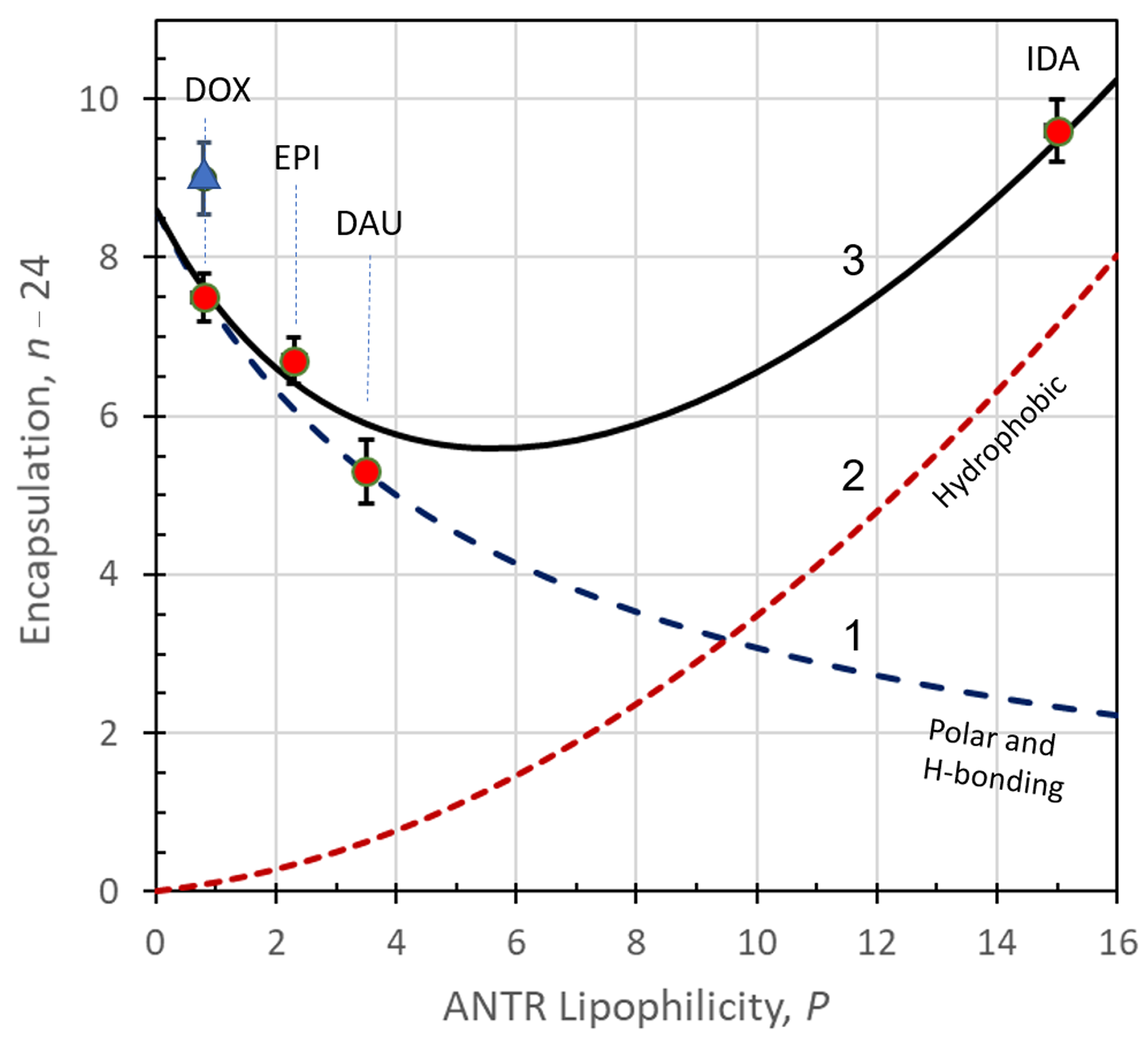
3.1. Mechanisms of Encapsulation of ANTR Drugs
3.2. Model Calculations for ANTR Drugs Encapsulation in APO Nanocages
- (1)
- ∆G0pol due to polar interactions leading to the formation of hydrogen bonds and immobilization of ANTR molecules on the inner surface of an APO cage on hydrophilic sites;
- (2)
- ∆G0hph due to hydrophobic interactions leading to the immobilization of ANTR on the inner APO cage surface on hydrophobic sites;
- (3)
- ∆G0ens due to the supramolecular ensemble formation APO(ANTRp), with the maximum encapsulation number P = nens,max = 24;
- (4)
- ∆G0trap due to the entrapment of free ANTR drug molecules in the inner volume of an APO nanocage;
- (5)
- ∆G0el due to electrostatic attraction of positively charged ANTR molecules, at pH < pKa of the protonation of the -NH2 moiety, and negatively charged dissociated local carboxyl groups of APO. The point of zero charge (pzc) for APO is: pzc = 4.4 [61,62]). The electrostatic interactions are mainly acting during the encapsulation and drug release processes, in acidic environments.
- (1)
- Binding of ANTR drugs on non-hydrophobic sites of APO.
CD (n1,max−n1)CAPO n1CAPO
- (i)
- a hyperbolic averaging for the number of sites lost with increasing P and the number of residual sites n1,res, which remain available even at the highest P due to the characteristic properties of the ANTR triple ring structure, or
- (ii)
- a loss function representing the number of blocked hydrophilic sites, which increases with increasing lipophilicity P, according to the phenomenological equation:
- (2)
- Binding of ANTR drugs on hydrophobic sites of APO
CD (n2,max − n2)CAPO n2CAPO
3.3. Performance of APO(ANTR) Nanodrugs in Treatment of HeLa and MDA-MB-231 Cancer Cell Lines and Non-Tumorigenic MCF10A Cells In Vitro
4. Materials and Methods
4.1. Chemicals
4.2. Apparatus
4.3. Software
4.4. Incorporation of Anthracycline Drugs to Apoferritin Nanocages
4.5. Modification of APO(Drug) Nanocages with Folic Acid
4.6. Encapsulation Ratio Drug:APO
4.7. Releasing of Anthracyclines form APO(Drug) Nanocages
4.8. Cell Culture
4.9. Cell Death Assay
4.10. Cytotoxicity/Growth Inhibition Tests by Crystal Violet Staining
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANTR | Anthracycline |
| APO | Apoferritin |
| CD | Circular dichroism |
| CDD | Controlled drug delivery |
| DAU | Daunorubicin |
| DOX | Doxorubicin |
| DMEM | Dulbecco’s Modified Eagle Medium |
| EDC | N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride |
| EGF | Epidermal growth factor |
| EPI | Epirubicin |
| FA | Folic acid |
| FBS | Fetal bovine serum |
| FR | Folate receptor |
| GSH | Glutathione |
| IDA | Idarubicin |
| MDR | Multiple drug resistance |
| MI buffer | McIlvain buffer |
| NHS | N-hydroxysuccinimide |
| NP | Nanoparticle |
| PBS | Phosphate-buffered saline |
| PCS | Percentage of cell survival |
| PMT | Photomultiplier tube |
| pzc | Point of zero charge |
| ROS | Reactive oxygen species |
| SCARA5 | Scavenger Receptor Class A Member 5 |
| TDD | Targeted drug delivery |
| TfR1 | Transferrin receptor 1 |
References
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760; [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.; Choi, S.K. Mechanisms of drug release in nanotherapeutic delivery systems. Chem Rev. 2015, 115, 3388–3432. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat Rev Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Kurzatkowska, K.; Santiago, T.; Hepel, M. Plasmonic nanocarrier grid-enhanced Raman sensor for studies of anticancer drug delivery. Biosens. Bioelectron. 2017, 91, 780–787. [Google Scholar] [CrossRef]
- Li, J.; Skeete, Z.; Shan, S.; Yan, S.; Kurzątkowska, K.; Zhao, W.; Ngo, Q.M.; Holubovska, P.; Luo, J.; Hepel, M.; et al. Surface enhanced Raman scattering detection of cancer biomarkers with bifunctional nanocomposite probe. Anal. Chem. 2015, 87, 10698–10702. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updat. 2015, 23, 69–78. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist. Updat. 2017, 31, 15–30. [Google Scholar] [CrossRef]
- Wielinga, P.R.; Westerhoff, H.V.; Lankelma, J. The relative importance of passive and P-glycoprotein mediated anthracycline efflux form multidrug-resistant cells. Eur. J. Biochem. 2000, 267, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Yhee, J.Y.; Jeon, S.; Yoon, H.Y.; Shim, M.K.; Ko, H.; Min, J.; Na, J.H.; Chang, H.; Han, H.; Kim, J.H.; et al. Effects of tumor microenvironments on targeted delivery of glycol chitosan nanoparticles. J. Control. Release 2017, 267, 223–231. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Son, S.; Lee, S.J.; You, D.G.; Yhee, J.Y.; Park, J.H.; Swierczewska, M.; Lee, S.; Kwon, I.C.; Kim, S.H.; et al. Glycol chitosan nanoparticles as specialized cancer therapeutic vehicles: Sequential delivery of doxorubicin and Bcl-2 siRNA. Sci. Rep. 2014, 4, 6878. [Google Scholar] [CrossRef] [Green Version]
- Grel, H.; Ratajczak, K.; Jakiela, S.; Stobiecka, M. Gated Resonance Energy Transfer (gRET) Controlled by Programmed Death Protein Ligand 1. Nanomaterials 2020, 10, 1592. [Google Scholar] [CrossRef]
- Ilkhani, H.; Hughes, T.; Li, J.; Zhong, C.J.; Hepel, M. Nanostructured SERS-electrochemical biosensors for testing of anticancer drug interactions with DNA. Biosens. Bioelectron. 2016, 80, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Trapani, G.; Denora, N.; Trapani, A.; Laquintana, V. Recent advances in ligand targeted therapy. J. Drug Targeting 2012, 20, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kue, C.S.; Kamkaew, A.; Burgess, K.; Kiew, L.V.; Chung, L.Y.; Lee, H.B. Small Molecules for Active Targeting in Cancer. Med. Res. Reviews 2016, 36, 494–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Di Penta, A.; Carmona, U.; Yang, F.; Schöps, R.; Brandsch, M.; Zugaza, J.L.; Knez, M. H-Chain Ferritin: A Natural Nuclei Targeting and Bioactive Delivery Nanovector. Adv. Healthcare Mater. 2015, 4, 1305–1310; [Google Scholar] [CrossRef] [PubMed]
- Santiago, T.; DeVaux, R.S.; Kurzątkowska, K.; Espinal, R.; Herschkowitz, J.I.; Hepel, M. Surface-enhanced Raman scattering investigation of targeted delivery and controlled release of gemcitabine. Int. J. Nanomed. 2017, 12, 7763–7776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Chen, M.; Wang, J. Nanocarriers with dual pH-sensitivity for enhanced tumor cell uptake and rapid intracellular drug release. RSC Adv. 2014, 4, 30780–30783. [Google Scholar] [CrossRef]
- Liang, M.; Fan, K.; Zhou, M.; Duan, S.; Zheng, J.; Yang, D.; Feng, J.; Yan, X. H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc. Natl. Acad. Sci. USA 2014, 111, 14900–14905; [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Bjorkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Hepel, M. Controlled release of targeted anti-leukemia drugs azacitidine and decitabine monitored using surface-enhanced Raman scattering (SERS) spectroscopy. Mediterr. J. Chem. 2017, 6, 125–132; [Google Scholar] [CrossRef]
- Running, L.; Espinal, R.; Hepel, M. Controlled release of targeted chemotherapeutic drug dabrafenib for melanoma cancers monitored using surface-enhanced Raman scattering (SERS) spectroscopy. Mediterr. J. Chem. 2018, 7, 18–27. [Google Scholar] [CrossRef]
- Hepel, M.; Stobiecka, M. Detection of Oxidative Stress Biomarkers Using Functional Gold Nanoparticles. In Fine Particles in Medicine and Pharmacy; Matijevic, E., Ed.; Springer Sci Publ.: New York, NY, USA, 2012; pp. 241–281. [Google Scholar]
- Stobiecka, M.; Hepel, M. Double-shell gold nanoparticle-based DNA-carriers with poly-L-lysine binding surface. Biomaterials 2011, 32, 3312–3321. [Google Scholar] [CrossRef]
- Hepel, M.; Stobiecka, M. Detection of Oxidative Stress Biomarkers Using Novel Nanostructured Biosensors. In New Perspectives in Biosensors Technology and Applications; Serra, P.A., Ed.; INTECH: Vienna, Austria, 2011; pp. 343–372. [Google Scholar] [CrossRef]
- Hepel, M.; Stobiecka, M.; Peachey, J.; Miller, J. Intervention of glutathione in pre-mutagenic catechol-mediated DNA damage in the presence of copper(II) ions. Mutat. Res. 2012, 735, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hepel, M.; Stobiecka, M. Comparative kinetic model of fluorescence enhancement in selective binding of monochlorobimane to glutathione. J. Photochem. Photobiol. A 2011, 225, 72–80. [Google Scholar] [CrossRef]
- Hepel, M.; Stobiecka, M. Supramolecular Interactions of Oxidative Stress Biomarker Glutathione with Fluorone Black. Spectrochim. Acta A 2018, 192, 146–152; [Google Scholar] [CrossRef] [PubMed]
- Truffi, M.; Fiandra, L.; Sorrentino, L.; Monieri, M.; Corsi, F.; Mazzucchelli, S. Ferritin nanocages: A biological platform for drug delivery, imaging and theranostics in cancer. Pharmacol. Res. 2016, 107, 57–65. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, X.; Diao, H.; Zhang, J.; Li, H.; Sun, H.; Guo, Z. Encapsulation of platinum anticancer drugs by apoferritin. Chem. Commun. 2007, 33, 3453–3455. [Google Scholar] [CrossRef]
- Kilic, M.; Ozlu, E.; Calies, S. A novel protein-based anticancer drug encapsulating nonosphere: Apoferritin-doxorubicin complex. J. Biomed. Nanotechnol. 2012, 8, 508–514. [Google Scholar] [CrossRef]
- Groot, A.S.D.; Scott, D.W. Immunogenicity of protein, therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef]
- Tosi, G.; Belletti, D.; Pederzoli, F.; Ruozi, B. Apoferritin nanocage as drug reservoir: Is it a reliable drug delivery system? Expert Opin. Drug Delivery 2016, 13, 1341–1343. [Google Scholar] [CrossRef] [Green Version]
- He, D.D.; Marles-Wright, J. Ferritin family proteins and their use in bionanotechnology. New Biotechnol. 2015, 32, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balletti, D.; Pederzoli, F.; Forni, F.; Vandelli, M.A.; Tosi, G.; Rouzi, B. Protein cage nanostructure as drug delivery system: Magnifying glass in appoferritin. Expert Opin. Drug Delivery 2017, 14, 825–840. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, K.; Lukasiak, A.; Grel, H.; Dworakowska, B.; Jakiela, S.; Stobiecka, M. Monitoring of dynamic ATP level changes by oligomycin-modulated ATP synthase inhibition in SW480 cancer cells using fluorescent “On-Off” switching DNA aptamer. Analytical and Bioanalytical Chemistry 2019, 411, 6899–6911; [Google Scholar] [CrossRef] [Green Version]
- Monti, M.; Ferraro, G.; Merlino, A. Ferritin-based anticancer metallodrug delivery: Crystallographic, analytical and cytotoxicity studies. Nanomed. Nanotechnol. Biol. Med. 2019, 20, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Wang, X.; Zhang, C.; Zhang, Y.; Wang, Q.; Yang, Z.; Guo, Z. Characterization and cellular uptake of platinum anticancer drugs encapsulated in apoferritin. J. Inorg. Biochem. 2009, 103, 1039–1044. [Google Scholar] [CrossRef]
- Pontillo, N.; Pane, F.; Messori, L.; Smoresano, A.; Merlino, A. Cisplatin encapsulation within a ferritin nanocage: A high-resolution crystallographic study. Chem. Commun. 2016, 52, 4136–4139. [Google Scholar] [CrossRef]
- Falvo, E.; Tremante, E.; Fraioli, R.; Leonetti, C.; Zamparelli, C.; Boffi, A.; Morea, V.; Ceci, P.; Giacomini, P. Anti-body conjugates: Targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale 2013, 5, 12278–12285. [Google Scholar] [CrossRef] [Green Version]
- Ruozi, B.; Veratti, P.; Vandelli, M.A.; Tombesi, A.; Tonelli, M.; Forni, F.; Pederzoli, F.; Belletti, D.; Tosi, G. Apoferritin nanocage as streptomycin drug reservoir: Technological optimization of a new drug delivery system. Int. J. Pharmac. 2017, 518, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wei, W.; Huang, S.; Lin, S.S.; Zhang, X.; Zhang, C.; Du, Y.; Ma, G.; Li, M.; Mann, S.; et al. Bio-inspired protein-gold nanoconstruct with core-void-shell structure: Beyond a chemo drug carrier. J. Mater. Chem. B 2013, 1, 3136–3143. [Google Scholar] [CrossRef]
- Ma-Ham, A.; Wu, H.; Wanh, J.; Kang, X.; Zhang, Y.; Lin, Y. Apoferritin-based nanomedicine platform for drug delivery: Equilibrium binding study of daunomycin with DNA. J. Mater. Chem. 2011, 21, 8700–8708. [Google Scholar] [CrossRef]
- Dostalova, S.; Cerna, T.; Hynek, D.; Koudelkova, Z.; Vaculovic, T.; Kopel, P.; Hrabeta, J.; Heger, Z.; Vaculovicova, M.; Eckschlager, T.; et al. Site-direct conjugation of antibodies to apoferritin nanocarrier for targeted drug delivery to prostate cancer cells. ACS Appl. Mater. Interfaces 2016, 8, 14430–14441. [Google Scholar] [CrossRef] [PubMed]
- Dostalova, S.; Vasickova, K.; Hynek, D.; Krizkova, S.; Richtera, L.; Vaculovicova, M.; Eckschlager, T.; Stiborova, M.; Heger, Z.; Adam, V. Apoferritin as ubiquitous nanocarrier with excellent shelf life. Int. J. Nanomed. 2017, 12, 2265–2278; [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhai, M.; Xie, X.; Zhang, Y.; Ma, S.; Li, Z.; Yu, F.; Zhao, B.; Zhang, M.; Yang, Y.; et al. Apoferritin nanocage for brain targeted doxorubicin delivery. Mol. Pharmaceutis 2017, 14, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Li, Z.; Yin, S.; Wang, Q.; Guo, F.; Zhang, Y.; Yu, R.; Liu, Y.; Su, Z. Extending half-life of H-ferritin nanoparticles by fusing albumin binding domain for doxorubicin encapsulation. Biomacromolecules 2018, 19, 773–781. [Google Scholar] [CrossRef]
- Alqaraghuli, H.G.J.; Kashanian, S.; Rafipour, R.; Mahdavian, E.; Mansouri, K. Develoopment and characterization of folic acid-functionalized apoferritin as a delivery vehicle for epirubicin against MCF-7 breasr cancer cells. Art. Cells Nanomed. Biotechnol. 2018, 46, 5847–5854; [Google Scholar] [CrossRef]
- Yan, F.; Zhang, Y.; Kim, K.S.; Yuan, H.K.; Vo-Dinh, T. Cellular uptake and photodynamic activity of protein nanocages containing methylene blue photosensitizing drug. Photochem. Photobiol. 2010, 86, 662–666. [Google Scholar] [CrossRef]
- Cutrin, J.C.; Crich, S.G.; Burghelea, D.; Dastrù, W.; Aime, S. Curcumin/Gd loaded apoferritin: A novel “theranostic” agent to prevent hepatocellular damage in toxic induced acute hepatitis. Mol. Pharm. 2013, 10, 2079–2085. [Google Scholar] [CrossRef]
- Tahover, E.; Patil, Y.P.; Gabizon, A.A. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: Focus on liposomes. Anti-Cancer Drugs 2015, 26, 241–258. [Google Scholar] [CrossRef]
- Hepel, M.; Stobiecka, M. Interactions of Herbicide Atrazine with DNA; Nova Science Publishers: New York, NY, USA, 2010; ISBN 978-1-61728-908-8. [Google Scholar]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol Rev. 2014, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early Detection of Anthracycline Cardiotoxicity and Improvement with Heart Failure Therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Jia, S.; Wang, Q.; Ding, X.; Liu, Y.; Yao, H.; Zhou, J. A self-targeting dual ROS/pH-responsive apoferritin nanocages for spatiotemporally controlled drug delivery to breast cancer. Biomacromolecules 2018, 19, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Hemati, M.; Haghiralsadat, F.; Jafary, F.; Moosavizadeh, S.; Moradi, A. Targeting cell cycle protein in gastric cancer with CDC20siRNA and anticancer drugs (doxorubicin and quercetin) co-loaded cationic PEGylated nanoniosomes. Int. J. Nanomed. 2019, 14, 6575–6585; [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wu, D.; Chen, Y.; Shan, G.; Liu, Y. Apoferritin nanocages with Au nanoshell coating as drug carrier for multistimuli-responsive drug release. Mater. Sci. Engng. C 2019, 95, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ke, J.; Zhou, X.E.; Yi, W.; Brunzelle, J.S.; Li, J.; Yong, E.L.; Xu, H.E.; Melcher, K. Structural basis for molecular recognition of folic acid by folate receptors. Nature 2013, 500, 486–489. [Google Scholar] [CrossRef] [Green Version]
- Husseini, G.; Kanan, S.; Al-Sayah, M. Investigating the fluorescence quenching of doxorubicin in folic acid solutions and its relation to ligand-targeted nanocarriers. J. Nanosci. Nanotechnol. 2016, 16, 1410–1414. [Google Scholar] [CrossRef]
- Santra, S.; Kaittanis, C.; Santiesteban, O.J.; Perez, J.M. Cell-Specific, Activatable, and theranostic prodrug for dual-targeted cancer imaging and therapy. J. Am. Chem. Soc. 2011, 133, 16680–16688. [Google Scholar] [CrossRef] [Green Version]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Mazur, A.; Litt, I.; Shorr, E. Chemical properties of ferritin and their relation to its vasodepressor activity. J. Biol. Chem. 1950, 187, 473–484. [Google Scholar] [CrossRef]
- Nakamura, T.; Konno, K. Studies on ferritin. J. Biochem. 1954, 41, 499–502. [Google Scholar] [CrossRef]
- Domínguez-Vera, J.M. Iron(III) complexation of Desferrioxamine B encapsulated in apoferritin. J. Inorg. Biochem. 2004, 98, 469–472. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anthracycline | Encapsulation Excess *, n (ANTR:APO) | Drug Lipophilicity ** Pdrug |
|---|---|---|
| Doxorubicin | 31.5 ± 0.6 | 0.8 ± 0.1 |
| Epirubicin | 30.7 ± 1.0 | 2.3 ± 0.3 |
| Daunorubicin | 29.3 ± 1.0 | 3.5 ± 0.5 |
| Idarubicin | 33.6 ± 0.7 | 15.0 ± 1.3 |
| ANTR Drug | EC50 for Free Drug nM | EC50 for APO(Drug) nM |
|---|---|---|
| Doxorubicin | 68.0 | 224.9 |
| Epirubicin | 39.8 | 289.8 |
| Idarubicin | 14.1 | 92.5 |
| Property | Inner APO Surface | Comments | Reference |
|---|---|---|---|
| APO surface area | 201.0 nm2 | rint = 4 nm, rext = 6 nm | this work |
| nDOX/APO, horizontal | 196 | theor., non-spec. ads. ** | this work |
| nDOX/APO, vertical | 308 | theor., non-spec. ads. | this work |
| nDOX/APO, side-on | 150 | theor., non-spec. ads. | this work |
| nDOX/APO, exprtl. | 24 ± 3 | APO-DOX complex | [30] |
| nDOX/APO, exprtl. | 33 *** | upload in 8 M urea | [18] |
| nDOX/APO, exprtl. | 31.5 ± 0.6 | − | this work |
| nIDA/APO, exprtl. | 33.6 ± 0.7 | IDA drug | this work |
| ncisPt/APO, exprtl. | 21.6–55.2 | cisplatin drug | [38] |
| ncisPt/APO, exprtl. | 15 | cisplatin drug | [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurzątkowska, K.; Pazos, M.A., II; Herschkowitz, J.I.; Hepel, M. Cancer-Targeted Controlled Delivery of Chemotherapeutic Anthracycline Derivatives Using Apoferritin Nanocage Carriers. Int. J. Mol. Sci. 2021, 22, 1362. https://doi.org/10.3390/ijms22031362
Kurzątkowska K, Pazos MA II, Herschkowitz JI, Hepel M. Cancer-Targeted Controlled Delivery of Chemotherapeutic Anthracycline Derivatives Using Apoferritin Nanocage Carriers. International Journal of Molecular Sciences. 2021; 22(3):1362. https://doi.org/10.3390/ijms22031362
Chicago/Turabian StyleKurzątkowska, Katarzyna, Manuel A. Pazos, II, Jason I. Herschkowitz, and Maria Hepel. 2021. "Cancer-Targeted Controlled Delivery of Chemotherapeutic Anthracycline Derivatives Using Apoferritin Nanocage Carriers" International Journal of Molecular Sciences 22, no. 3: 1362. https://doi.org/10.3390/ijms22031362