Prediction of Protein–Protein Binding Interactions in Dimeric Coiled Coils by Information Contained in Folding Energy Landscapes



Abstract
:1. Introduction
2. Results
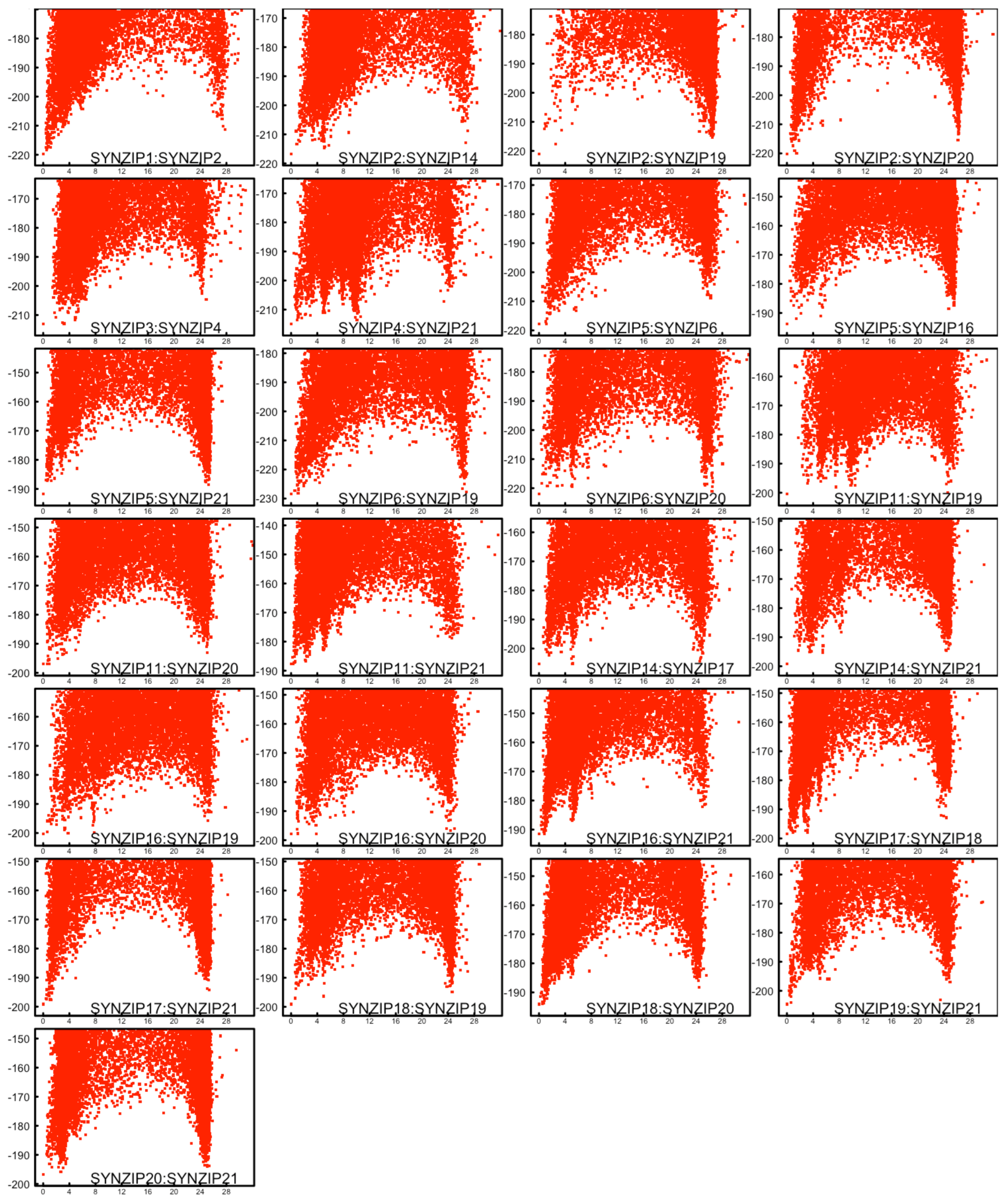
2.1. Modelling Folding Energy Landscapes of Dimeric Coiled Coils
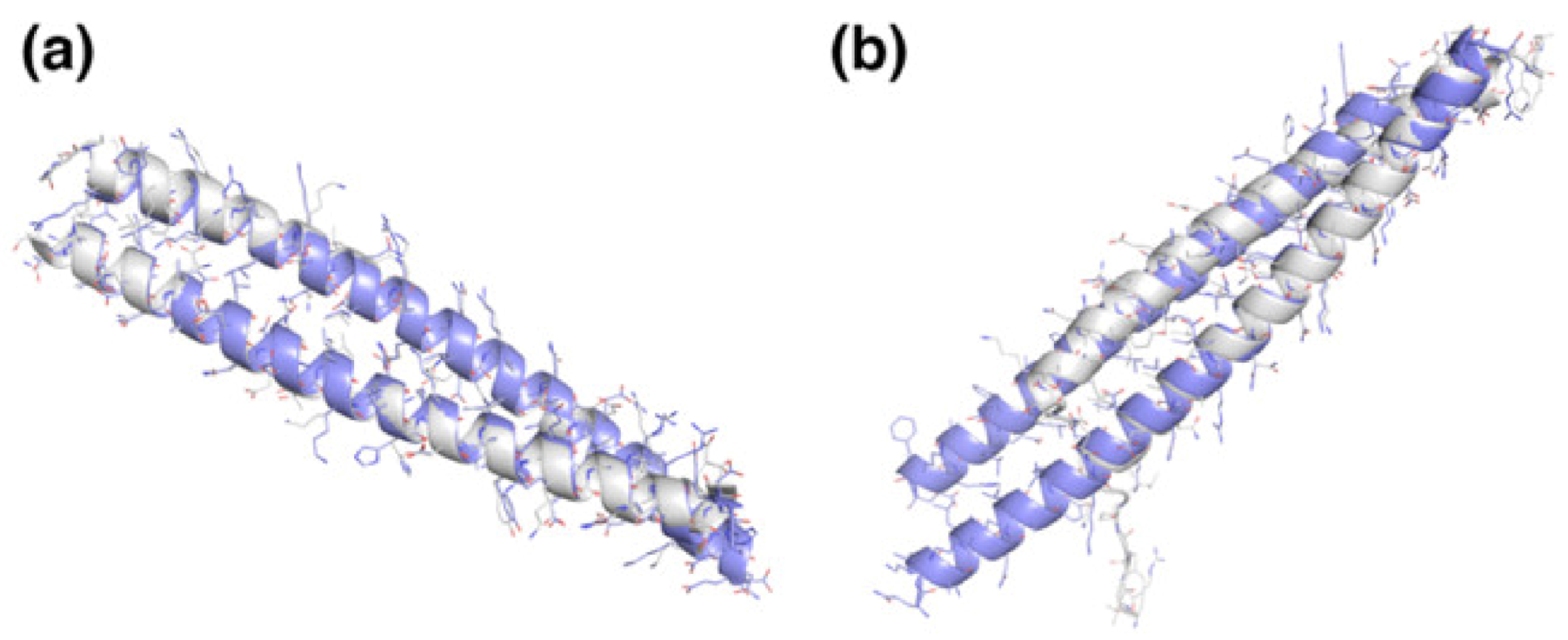
2.2. Energetics of the Highest Affinity Coiled Coil Dimers and Comparison to Experimental Structures
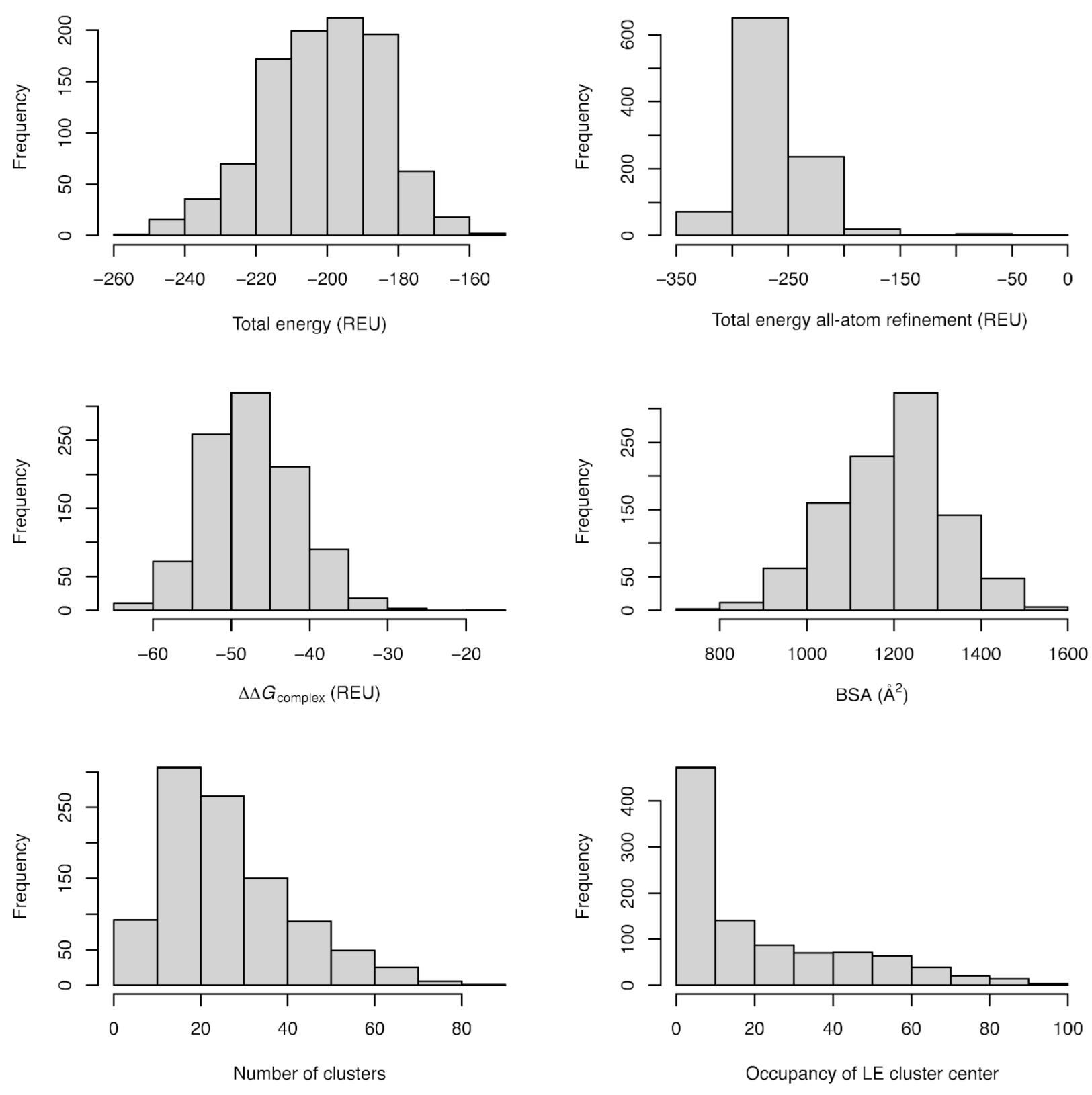
2.3. Characteristics of the Energy Landscapes and Properties of the Lowest Energy Models
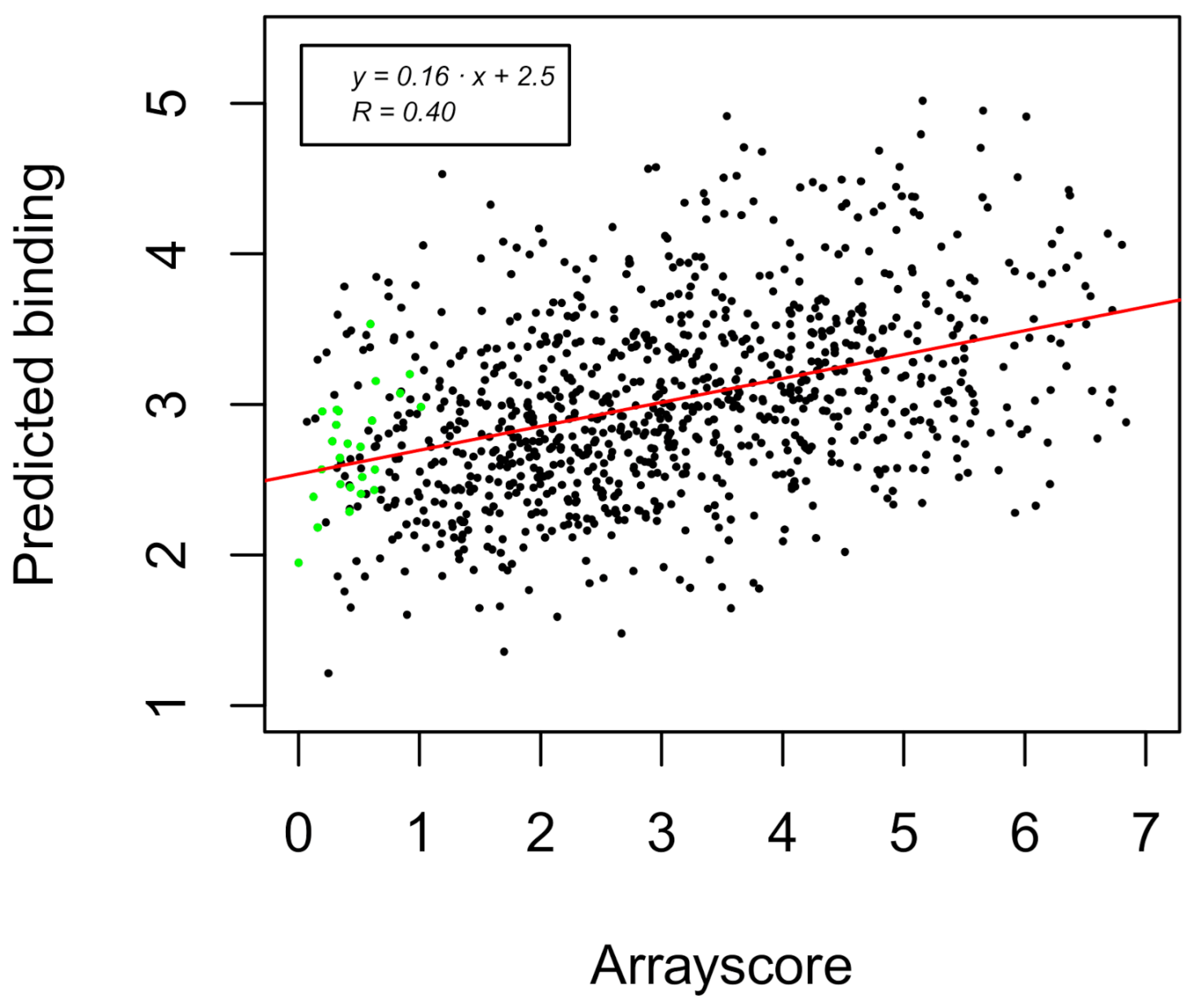
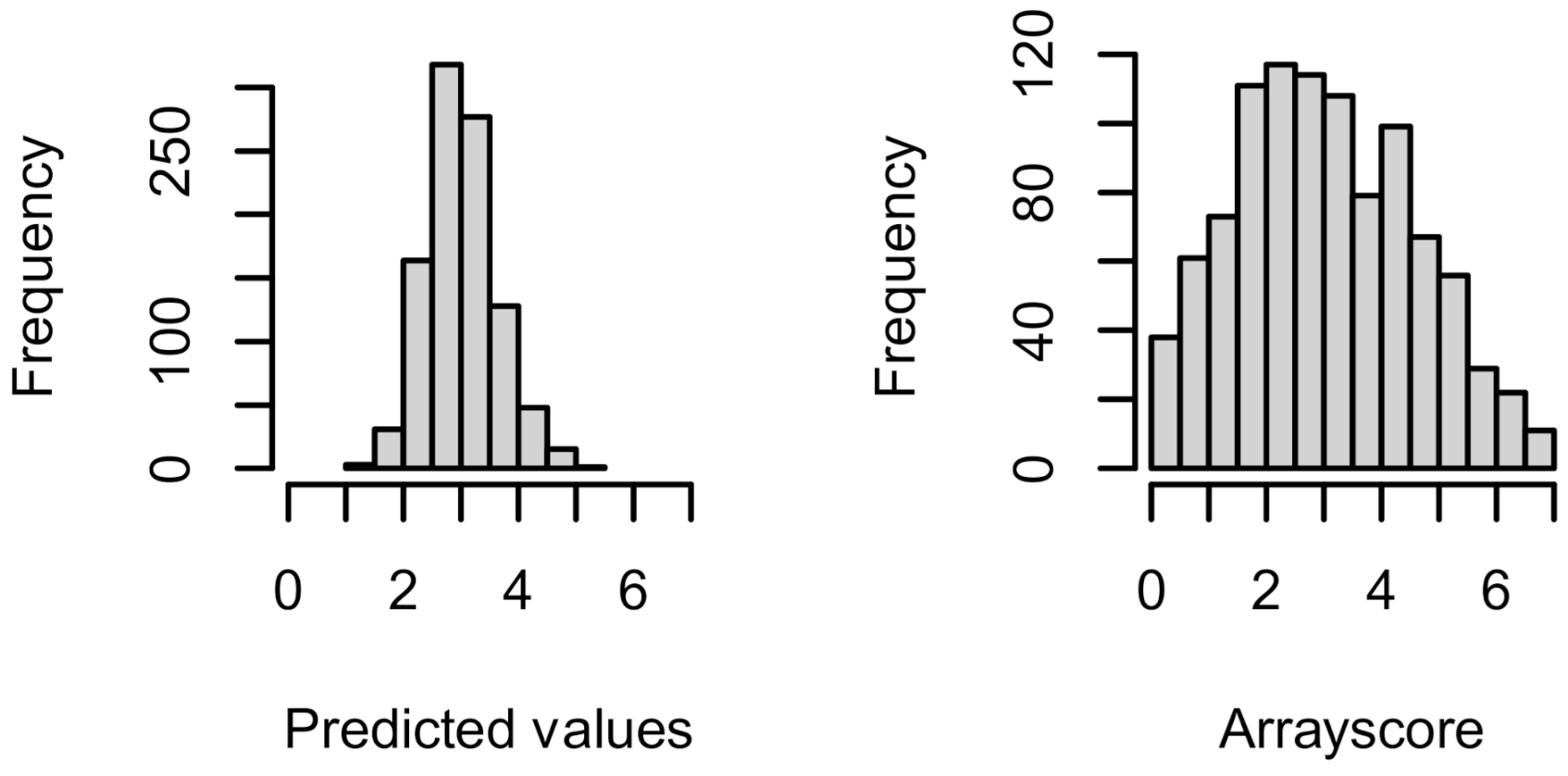
2.4. Prediction of the Coiled Coil Binding Interactions
3. Discussion
4. Materials and Methods
4.1. Coiled Coil Data Set
4.2. Estimate of the Coiled Coil Dimer Interaction
4.3. Computational Folding Simulations
4.4. Clustering, Energy Refinement, and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Buried surface area |
| REU | Rosetta energy units |
References
- Burkhard, P.; Stetefeld, J.; Strelkov, S.V. Coiled Coils: A Highly Versatile Protein Folding Motif. Trends Cell Biol. 2001, 11, 82–88. [Google Scholar] [CrossRef]
- Crick, F.H. Is α-Keratin a Coiled Coil? Nature 1952, 170, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rost, B. Comparing Function and Structure between Entire Proteomes. Protein Sci. 2001, 10, 1970–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crick, F. The Packing of α-Helices: Simple Coiled-Coils. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.A. Coiled-Coils in α-Helix-Containing Proteins: Analysis of the Residue Types within the Heptad Repeat and the Use of These Data in the Prediction of Coiled-Coils in Other Proteins. Biosci. Rep. 1982, 2, 1017–1024. [Google Scholar] [PubMed]
- Lupas, A. Coiled Coils: New Structures and New Functions. Trends Biochem. Sci. 1996, 21, 375–382. [Google Scholar] [CrossRef]
- Cohen, C.; Parry, D.A. α-Helical Coiled Coils and Bundles: How to Design an α-Helical Protein. Proteins Struct. Funct. Bioinform. 1990, 7, 1–15. [Google Scholar] [CrossRef]
- Gruber, M.; Soding, J.; Lupas, A.N. Comparative Analysis of Coiled-Coil Prediction Methods. J. Struct. Biol. 2006, 155, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Gradisar, H.; Bozic, S.; Doles, T.; Vengust, D.; Hafner-Bratkovic, I.; Mertelj, A.; Webb, B.; Sali, A.; Klavzar, S.; Jerala, R. Design of a Single-Chain Polypeptide Tetrahedron Assembled from Coiled-Coil Segments. Nat. Chem. Biol. 2013, 9, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Grigoryan, G.; Kim, Y.H.; Acharya, R.; Axelrod, K.; Jain, R.M.; Willis, L.; Drndic, M.; Kikkawa, J.M.; DeGrado, W.F. Computational Design of Virus-Like Protein Assemblies on Carbon Nanotube Surfaces. Science 2011, 332, 1071–1076. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.S.; Oberdorfer, G.; Xu, C.; Pei, X.Y.; Nannenga, B.L.; Rogers, J.M.; DiMaio, F.; Gonen, T.; Luisi, B.; Baker, D. High Thermodynamic Stability of Parametrically Designed Helical Bundles. Science 2014, 346, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccai, N.R.; Chi, B.; Thomson, A.R.; Boyle, A.L.; Bartlett, G.J.; Bruning, M.; Linden, N.; Sessions, R.B.; Booth, P.J.; Brady, R.L.; et al. A De Novo Peptide Hexamer with a Mutable Channel. Nat. Chem. Biol. 2011, 7, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Andre, I.; Shen, Y.; Wu, Y.; Lemak, A.; Bansal, S.; Arrowsmith, C.H.; Szyperski, T.; Baker, D. Simultaneous Prediction of Protein Folding and Docking at High Resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 18978–18983. [Google Scholar] [CrossRef] [Green Version]
- Ramisch, S.; Lizatovic, R.; Andre, I. Exploring Alternate States and Oligomerization Preferences of Coiled-Coils by De Novo Structure Modeling. Proteins 2015, 83, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Tyka, M.D.; Keedy, D.A.; Andre, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Andre, I.; Bjelic, S. Computational Assessment of Folding Energy Landscapes in Heterodimeric Coiled Coils. Proteins 2018, 86, 790–801. [Google Scholar] [CrossRef]
- Reinke, A.W.; Grant, R.A.; Keating, A.E. A Synthetic Coiled-Coil Interactome Provides Heterospecific Modules for Molecular Engineering. J. Am. Chem. Soc. 2010, 132, 6025–6031. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.E.; Bashor, C.J.; Lim, W.A.; Keating, A.E. SYNZIP Protein Interaction Toolbox: In Vitro and In Vivo Specifications of Heterospecific Coiled-Coil Interaction Domains. ACS Synth. Biol. 2012, 1, 118–129. [Google Scholar] [CrossRef]
- Chen, J.; Sawyer, N.; Regan, L. Protein-Protein Interactions: General Trends in the Relationship between Binding Affinity and Interfacial Buried Surface Area. Protein Sci. 2013, 22, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Benedix, A.; Becker, C.M.; de Groot, B.L.; Caflisch, A.; Bockmann, R.A. Predicting Free Energy Changes Using Structural Ensembles. Nat. Methods 2009, 6, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potapov, V.; Kaplan, J.B.; Keating, A.E. Data-Driven Prediction and Design of bZIP Coiled-Coil Interactions. PLoS Comput. Biol. 2015, 11, e1004046. [Google Scholar] [CrossRef] [PubMed]
- Shringari, S.R.; Giannakoulias, S.; Ferrie, J.J.; Petersson, E.J. Rosetta Custom Score Functions Accurately Predict DeltaDeltaG of Mutations at Protein-Protein Interfaces Using Machine Learning. Chem. Commun. 2020, 56, 6774–6777. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-Based Prediction of Binding Affinity in Protein-Protein Complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Alm, E.; Baker, D. Prediction of Protein-Folding Mechanisms from Free-Energy Landscapes Derived from Native Structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11305–11310. [Google Scholar] [CrossRef] [Green Version]
- Chavez, L.L.; Onuchic, J.N.; Clementi, C. Quantifying the Roughness on the Free Energy Landscape: Entropic Bottlenecks and Protein Folding Rates. J. Am. Chem. Soc. 2004, 126, 8426–8432. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Baker, D. Role of the Biomolecular Energy Gap in Protein Design, Structure, and Evolution. Cell 2012, 149, 262–273. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Schueler-Furman, O. Funnel Hunting in a Rough Terrain: Learning and Discriminating Native Energy Funnels. Structure 2008, 16, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Plaxco, K.W.; Simons, K.T.; Baker, D. Contact Order, Transition State Placement and the Refolding Rates of Single Domain Proteins. J. Mol. Biol. 1998, 277, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Rao, F.; Caflisch, A. The Protein Folding Network. J. Mol. Biol. 2004, 342, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. The Pymol Molecular Graphics System. Version 2.4.0. 2002. Available online: www.pymol.org (accessed on 22 January 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coiled Coil Dimer | Kd (nM) | Coiled Coil Dimer | Kd (nM) |
|---|---|---|---|
| SYNZIP1:SYNZIP2 | <10 | SYNZIP11:SYNZIP21 | <10 |
| SYNZIP2:SYNZIP14 | <10 | SYNZIP14:SYNZIP17 | <10 |
| SYNZIP2:SYNZIP19 | <10 | SYNZIP14:SYNZIP21 | <10 |
| SYNZIP2:SYNZIP20 | <10 | SYNZIP16:SYNZIP19 | <10 |
| SYNZIP3:SYNZIP4 | <30 | SYNZIP16:SYNZIP20 | <10 |
| SYNZIP4:SYNZIP21 | <10 | SYNZIP16:SYNZIP21 | <10 |
| SYNZIP5:SYNZIP6 | <15 | SYNZIP17:SYNZIP18 | <10 |
| SYNZIP5:SYNZIP16 | <10 | SYNZIP17:SYNZIP21 | <10 |
| SYNZIP5:SYNZIP21 | <10 | SYNZIP18:SYNZIP19 | <10 |
| SYNZIP6:SYNZIP19 | <10 | SYNZIP18:SYNZIP20 | <15 |
| SYNZIP6:SYNZIP20 | <10 | SYNZIP19:SYNZIP21 | <10 |
| SYNZIP11:SYNZIP19 | <10 | SYNZIP20:SYNZIP21 | <10 |
| SYNZIP11:SYNZIP20 | <10 |
| Variable | Variable Name | RPearson (Pred. vs. Exp) |
|---|---|---|
| x1 | Total energy | 0.19 |
| x2 | ∆∆Gcomplex | 0.27 |
| x3 | Number of clusters | 0.32 |
| x1 + x2 | 0.27 | |
| x1 + x3 | 0.39 | |
| x2 + x3 | 0.38 | |
| x1 + x2 + x3 | 0.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgoulia, P.S.; Bjelic, S. Prediction of Protein–Protein Binding Interactions in Dimeric Coiled Coils by Information Contained in Folding Energy Landscapes. Int. J. Mol. Sci. 2021, 22, 1368. https://doi.org/10.3390/ijms22031368
Georgoulia PS, Bjelic S. Prediction of Protein–Protein Binding Interactions in Dimeric Coiled Coils by Information Contained in Folding Energy Landscapes. International Journal of Molecular Sciences. 2021; 22(3):1368. https://doi.org/10.3390/ijms22031368
Chicago/Turabian StyleGeorgoulia, Panagiota S., and Sinisa Bjelic. 2021. "Prediction of Protein–Protein Binding Interactions in Dimeric Coiled Coils by Information Contained in Folding Energy Landscapes" International Journal of Molecular Sciences 22, no. 3: 1368. https://doi.org/10.3390/ijms22031368