
CD44 and Tumor-Derived Extracellular Vesicles (TEVs). Possible Gateway to Cancer Metastasis


Abstract
:1. Introduction
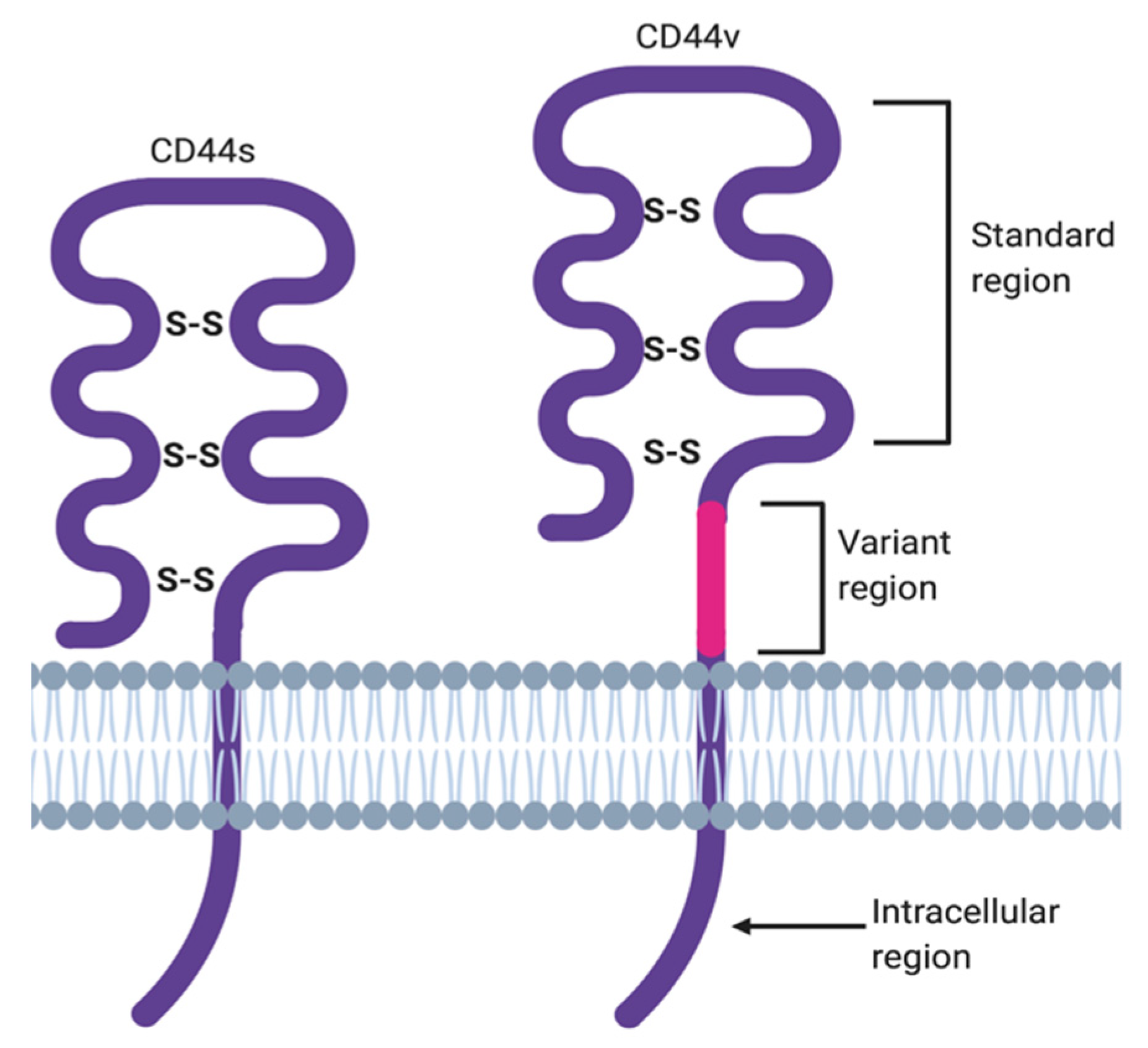
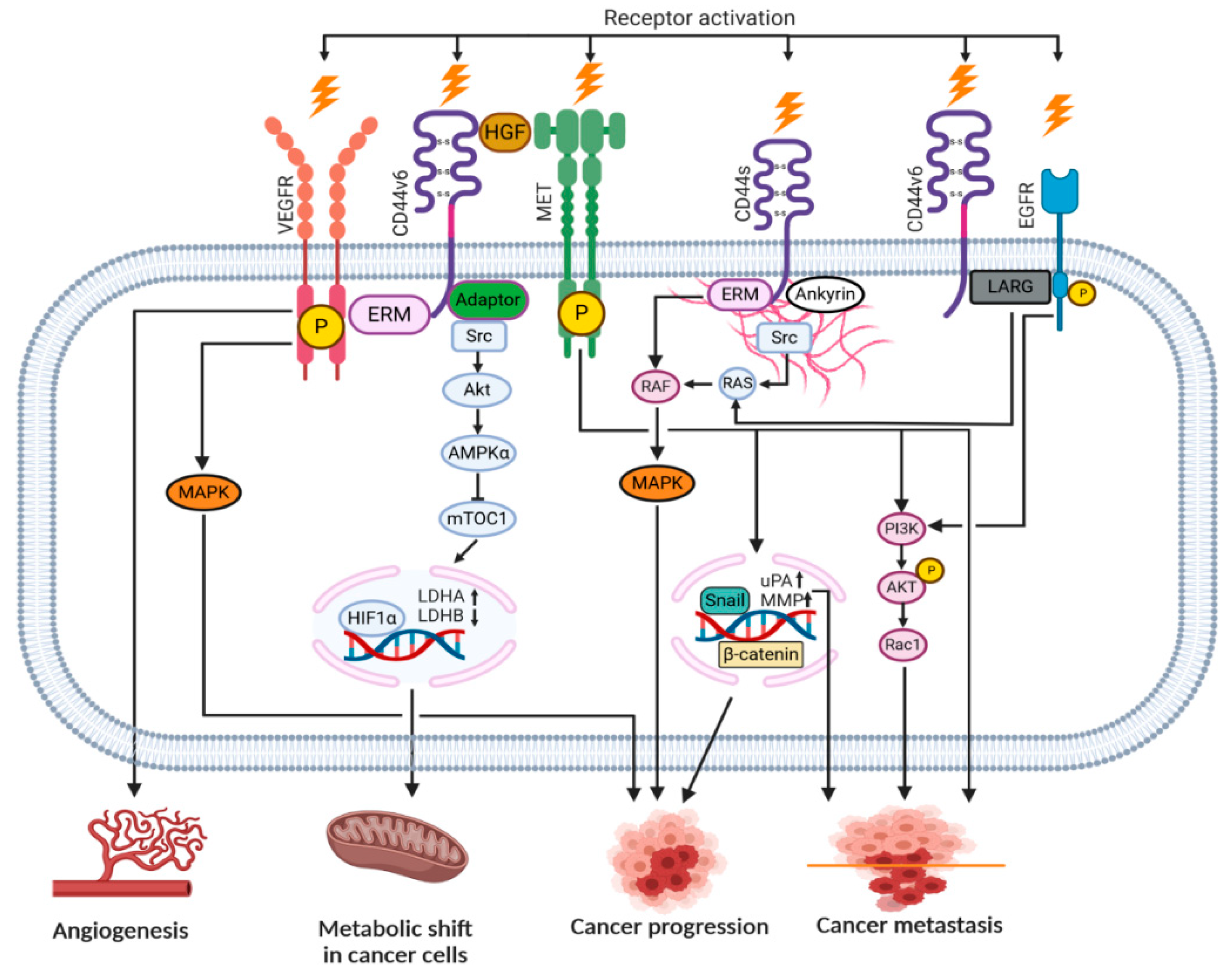
2. ‘Facts Check’—CD44 Receptor
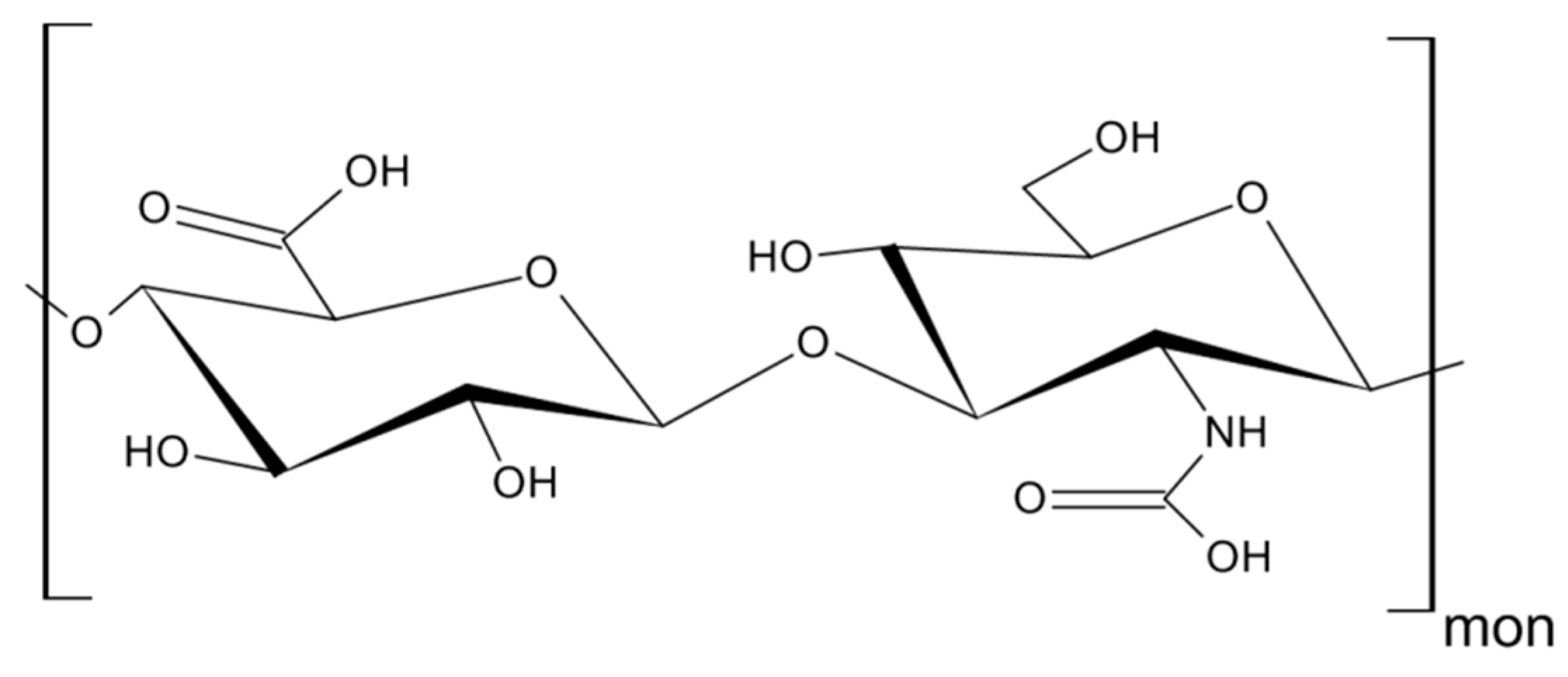
3. Hyaluronan—The Major Ligand of CD44
4. CD44-HA Interactions
5. EMT and the Role of CD44 and TEVs
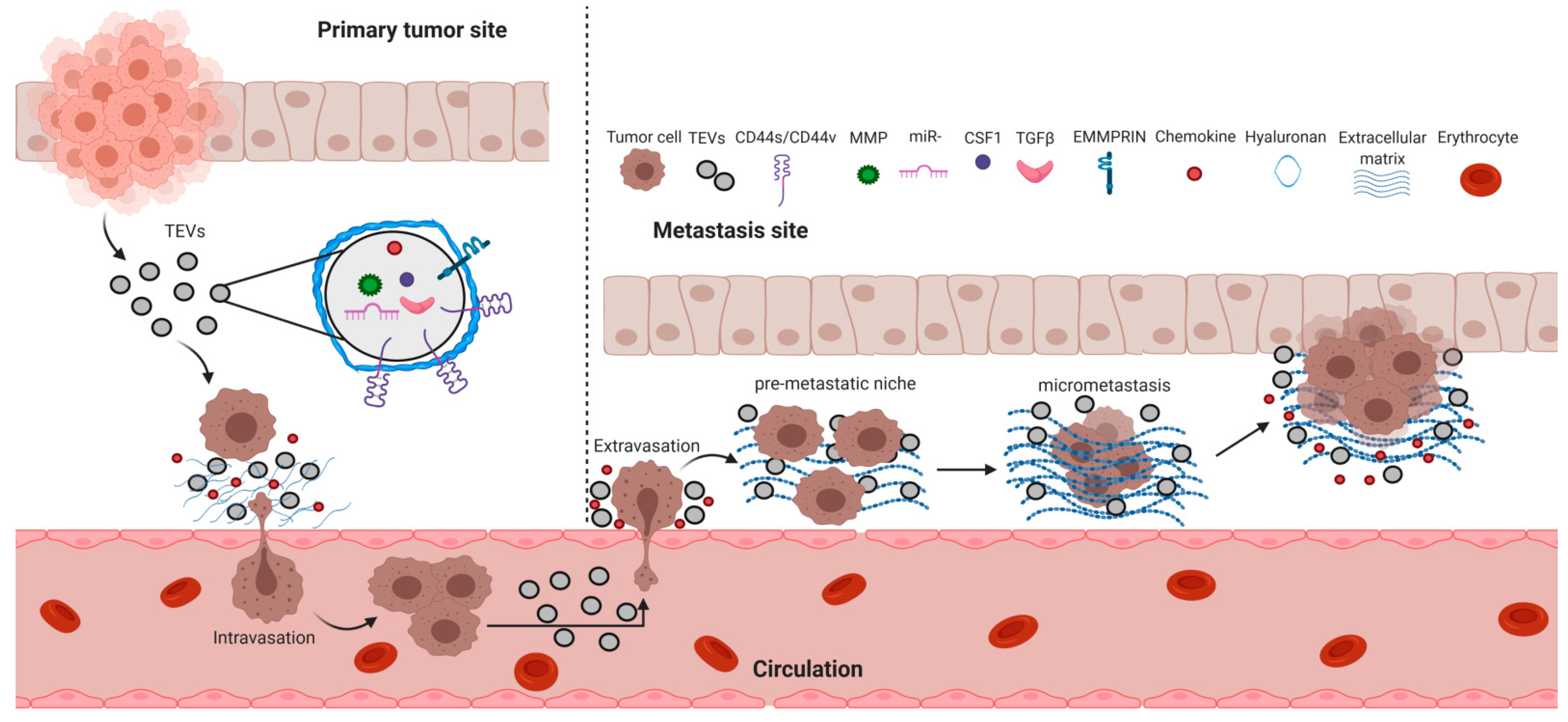
6. Cancer Metastasis and TEVs
6.1. Tumor Cell Intravasation
6.2. Tumor Cell Extravasation
6.3. Tumor Latency
6.4. Pre-Metastatic Niche Formation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TEVs | tumor-derived extracellular vesicles |
| EVs | extracellular vesicles |
| CD44 | cluster of differentiation 44 |
| CD44s | cluster of differentiation 44 standard isoform |
| CD44v | cluster of differentiation 44 variant isoform |
| HA | hyaluronan |
| HAS | hyaluronan synthase |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transformation |
| RTK | receptor tyrosine kinase |
| MMP | matrix matalloproteinase |
| CIC | cancer initiating cell |
| EMMPRIN | extracellular matrix metalloproteinase inducer |
| EGFR | epidermal growth factor receptor |
| CAFs | cancer associated fibroblasts |
| CRC | colorectal carcinoma |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| ERM | ezrin-radixin-moesin |
| MAPK | mitogen-activated protein kinase |
| PI3K | phosphoinositide 3-kinase |
| AKT | alpha serine/threonine-protein kinase |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| ICD | intracytoplasmic domain |
| RHAMM | receptor for hyaluronan-mediated motility |
| cSrc | proto-oncogene tyrosine-protein kinase Src |
| RAS | proto-oncogene protein p21 |
| FAK | focal adhesion kinase |
| Met | hepatocyte growth factor receptor |
| Snail | zinc finger protein SNAI1 |
References
- Yuana, Y.; Böing, A.N.; Grootemaat, A.E.; van der Pol, E.; Hau, C.M.; Cizmar, P.; Buhr, E.; Sturk, A.; Nieuwland, R. Handling and storage of human body fluids for analysis of extracellular vesicles. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38. [Google Scholar] [CrossRef]
- Wu, K.; Xing, F.; Wu, S.Y.; Watabe, K. Extracellular vesicles as emerging targets in cancer: Recent development from bench to bedside. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 538–563. [Google Scholar] [CrossRef]
- Härkönen, K.; Oikari, S.; Kyykallio, H.; Capra, J.; Hakkola, S.; Ketola, K.; Thanigai Arasu, U.; Daaboul, G.; Malloy, A.; Oliveira, C.; et al. CD44s assembles hyaluronan coat on filopodia and extracellular vesicles and induces tumorigenicity of MKN74 gastric carcinoma cells. Cells 2019, 276. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Jung, T.; Castellana, D.; Klingbeil, P.; Hernández, I.C.; Vitacolonna, M.; Orlicky, D.J.; Roffler, S.R.; Brodt, P.; Zöller, M. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 2009, 11. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Rana, S.; Zöller, M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia (US) 2013, 15. [Google Scholar] [CrossRef] [Green Version]
- Cichy, J.; Puré, E. The liberation of CD44. J. Cell Biol. 2003, 161, 839–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zuo, X.; Wei, D. Concise review: Emerging role of CD44 in cancer stem cells: A promising biomarker and therapeutic target. Stem Cells Transl. Med. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Sneath, R.J.S.; Mangham, D.C. The normal structure and function of CD44 and its role in neoplasia. J. Clin. Pathol. Mol. Pathol. 1998, 51, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Voort, R.; Taher, T.E.I.; Wielenga, V.J.M.; Spaargaren, M.; Prevo, R.; Smit, L.; David, G.; Hartmann, G.; Gherardi, E.; Pals, S.T. Heparan sulfate-modified CD44 promotes hepatocyte growth factor/scatter factor-induced signal transduction through the receptor tyro sine kinase c-Met. J. Biol. Chem. 1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Wen, T.; Ge, Y.; Liu, J.; Yang, L.; Jiang, Y.; Dong, X.; Liu, H.; Yao, J.; An, G. Disruption of Core 1-mediated O-glycosylation oppositely regulates CD44 expression in human colon cancer cells and tumor-derived exosomes. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cichy, J.; Puré, E. Cytokines regulate the affinity of soluble CD44 for hyaluronan. FEBS Lett. 2004. [Google Scholar] [CrossRef] [Green Version]
- Cichy, J.; Puré, E. Oncostatin M and transforming growth factor-β1 induce post-translational modification and hyaluronan binding to CD44 in lung-derived epithelial tumor cells. J. Biol. Chem. 2000. [Google Scholar] [CrossRef] [Green Version]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, function, and association with the malignant process. Adv. Cancer Res. 1997, 71, 241–319. [Google Scholar]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Yu, Q.; Toole, B.P. A new alternatively spliced exon between v9 and v10 provides a molecular basis for synthesis of soluble CD44. J. Biol. Chem. 1996. [Google Scholar] [CrossRef] [Green Version]
- Katoh, S.; McCarthy, J.B.; Kincade, P.W. Characterization of soluble CD44 in the circulation of mice: Levels are affected by immune activity and tumor growth. J. Immunol. 1994, 153. [Google Scholar]
- Katoh, S.; Taniguchi, H.; Matsubara, Y.; Matsumoto, N.; Fukushima, K.; Kadota, J.; Matsukura, S.; Kohno, S. Overexpression of CD44 on alveolar eosinophils with high concentrations of soluble CD44 in bronchoalveolar lavage fluid in patients with eosinophilic pneumonia. Allergy Eur. J. Allergy Clin. Immunol. 1999, 54. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Dennis, K.; Peschon, J.J.; Chandrasekaran, R.; Mikecz, K. Antibody-induced shedding of CD44 from adherent cells is linked to the assembly of the cytoskeleton. J. Immunol. 2001, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Päll, T.; Pink, A.; Kasak, L.; Turkina, M.; Anderson, W.; Valkna, A.; Kogerman, P. Soluble CD44 interacts with intermediate filament protein vimentin on endothelial cell surface. PLoS ONE 2011, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schwertfeger, K.L.; Cowman, M.K.; Telmer, P.G.; Turley, E.A.; McCarthy, J.B. Hyaluronan, inflammation, and breast cancer progression. Front. Immunol. 2015, 6, 236. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan-CD44 interactions in cancer: Paradoxes and possibilities. Clin. Cancer Res. 2009, 15, 7462–7468. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 2011, 91, 221–264. [Google Scholar] [CrossRef] [Green Version]
- Tolg, C.; McCarthy, J.B.; Yazdani, A.; Turley, E.A. Hyaluronan and RHAMM in Wound Repair and the “cancerization” of Stromal Tissues. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- McKee, C.M.; Penno, M.B.; Cowman, M.; Burdick, M.D.; Strieter, R.M.; Bao, C.; Noble, P.W. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages: The role of HA size and CD44. J. Clin. Invest. 1996. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007. [Google Scholar] [CrossRef]
- Ruffell, B.; Johnson, P. Hyaluronan induces cell death in activated T cells through CD44. J. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auvinen, P.; Rilla, K.; Tumelius, R.; Tammi, M.; Sironen, R.; Soini, Y.; Kosma, V.M.; Mannermaa, A.; Viikari, J.; Tammi, R. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res. Treat. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lokeshwar, V.B.; Öbek, C.; Soloway, M.S.; Block, N.L. Tumor-associated hyaluronic acid: A new sensitive and specific urine marker for bladder cancer. Cancer Res. 1997, 57, 773–777. [Google Scholar] [PubMed]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikström, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ropponen, K.; Tammi, M.; Parkkinen, J.; Eskelinen, M.; Tammi, R.; Lipponen, P.; Ågren, U.; Alhava, E.; Kosma, V.M. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 1998, 58, 342–347. [Google Scholar] [PubMed]
- McBride, W.H.; Bard, J.B.L. Hyaluronidase-sensitive halos around adherent cells: Their role in blocking lymphocyte-mediated cytolysis. J. Exp. Med. 1979. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Hirata, T.; Hayasaka, H.; Stern, R.; Murai, T.; Miyasaka, M. Tumor cells enhance their own CD44 cleavage and motility by generating hyaluronan fragments. J. Biol. Chem. 2006. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Murai, T.; Nishinakamura, H.; Kawashima, H.; Saya, H.; Miyasaka, M. Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J. Biol. Chem. 2003. [Google Scholar] [CrossRef] [Green Version]
- Tzircotis, G.; Thorne, R.F.; Isacke, C.M. Chemotaxis towards hyaluronan is dependent on CD44 expression and modulated by cell type variation in CD44-hyaluronan binding. J. Cell Sci. 2005. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sayer, S.S.M.; Dong, Y.; Arif, A.A.; Olsson, M.; Brown, K.L.; Johnson, P. The where, when, how and why of hyaluronan binding by immune cells. Front. Immunol. 2015, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Bennett, K.L.; Modrell, B.; Greenfield, B.; Bartolazzi, A.; Stamenkovic, I.; Peach, R.; Jackson, D.G.; Spring, F.; Aruffo, A. Regulation of CD44 binding to hyaluronan by glycosylation of variably spliced exons. J. Cell Biol. 1995. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.S.; Alves, C.S.; Wirtz, D.; Konstantopoulos, K. Distinct kinetic and molecular requirements govern CD44 binding to hyaluronan versus fibrin(ogen). Biophys. J. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, G.M.; Avenoso, A.; Micali, A.; Nastasi, G.; Squadrito, F.; Altavilla, D.; Bitto, A.; Polito, F.; Rinaldi, M.G.; Calatroni, A.; et al. High-molecular weight hyaluronan reduced renal PKC activation in genetically diabetic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesley, J.; Hascall, V.C.; Tammi, M.; Hyman, R. Hyaluronan binding by cell surface CD44. J. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010, 70. [Google Scholar] [CrossRef] [Green Version]
- Termeer, C.; Sleeman, J.P.; Simon, J.C. Hyaluronan—Magic glue for the regulation of the immune response? Trends Immunol. 2003, 24, 112–114. [Google Scholar] [CrossRef]
- Baaten, B.J.G.; Li, C.R.; Bradley, L.M. Multifaceted regulation of T cells by CD44. Commun. Integr. Biol. 2010, 3. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.; Oh, S.; Shin, I. Ablation of CD44 induces glycolysis-to-oxidative phosphorylation transition via modulation of the c-Src-Akt-LKB1-AMPK pathway. Biochem. J. 2016, 473, 3013–3030. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.S.; Oh, S.; Lee, K.M.; Yoo, S.A.; Shin, I. CD44 regulates cell proliferation, migration, and invasion via modulation of c-Src transcription in human breast cancer cells. Cell. Signal. 2015, 27. [Google Scholar] [CrossRef]
- Lokeshwar, V.B.; Mirza, S.; Jordan, A. Targeting hyaluronic acid family for cancer chemoprevention and therapy. Adv. Cancer Res. 2014, 123, 35–65. [Google Scholar] [PubMed] [Green Version]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitovic, D.; Kouvidi, K.; Karamanos, N.K.; Tzanakakis, G.N. The roles of hyaluronan/RHAMM/CD44 and their respective interactions along the insidious pathways of fibrosarcoma progression. BioMed Res. Int. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Morrison, H.; Matzke, A.; Kastilan, T.; Pace, G.; Herrlich, P.; Ponta, H. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-actin. Mol. Biol. Cell 2007, 18. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinke, L.M.; Xu, Y.; Cheng, C. Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baj-Krzyworzeka, M.; Weglarczyk, K.; Szatanek, R.; Mytar, B.; Baran, J.; Siedlar, M. The role of CD44H molecule in the interactions between human monocytes and pancreatic adenocarcinoma-derived microvesicles. Folia Histochem. Cytobiol. 2019, 57. [Google Scholar] [CrossRef]
- Cho, S.H.; Park, Y.S.; Kim, H.J.; Kim, C.H.; Lim, S.W.; Huh, J.W.; Lee, J.H.; Kim, H.R. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int. J. Oncol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Toole, B.P.; Zoltan-Jones, A.; Misra, S.; Ghatak, S. Hyaluronan: A critical component of epithelial-mesenchymal and epithelial-carcinoma transitions. Cells Tissues Organs 2005. [Google Scholar] [CrossRef]
- Kletukhina, S.; Neustroeva, O.; James, V.; Rizvanov, A.; Gomzikova, M. Role of mesenchymal stem cell-derived extracellular vesicles in epithelial-mesenchymal transition. Int. J. Mol. Sci. 2019, 20, 4813. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Tolg, C.; Turley, E. Dissecting the dual nature of hyaluronan in the tumor microenvironment. Front. Immunol. 2019, 10, 947. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.K.; Greening, D.W.; Rai, A.; Chen, M.; Xu, R.; Shafiq, A.; Mathias, R.A.; Zhu, H.J.; Simpson, R.J. Extracellular vesicles: Their role in cancer biology and epithelial-mesenchymal transition. Biochem. J. 2017, 474, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Franzen, C.A.; Blackwell, R.H.; Todorovic, V.; Greco, K.A.; Foreman, K.E.; Flanigan, R.C.; Kuo, P.C.; Gupta, G.N. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Barger, J.F.; Lovat, F.; Gao, M.; Otterson, G.A.; Nana-Sinkam, P. Lung cancer exosomes as drivers of epithelial mesenchymal transition. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, X.; Wang, J.; Li, M.; Cao, C.; Tan, J.; Ma, D.; Gao, Q. TGFβ1 in fibroblasts-derived exosomes promotes epithelialmesenchymal transition of ovarian cancer cells. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Szatanek, R.; Weglarczyk, K.; Stec, M.; Baran, J.; Parlinska-Wojtan, M.; Siedlar, M.; Baj-Krzyworzeka, M. Autologous tumor-derived microvesicles influence gene expression profiles and enhance protumorigenic chemotactic potential, signal transduction and cellular respiration in gastric cancer cells. Int. J. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sawada, K.; Kinose, Y.; Yoshimura, A.; Toda, A.; Nakatsuka, E.; Hashimoto, K.; Mabuchi, S.; Morishige, K.I.; Kurachi, H.; et al. Exosomes promote ovarian cancer cell invasion through transfer of CD44 to peritoneal mesothelial cells. Mol. Cancer Res. 2017, 15. [Google Scholar] [CrossRef] [Green Version]
- Baj-Krzyworzeka, M.; Szatanek, R.; Wȩglarczyk, K.; Baran, J.; Urbanowicz, B.; Brański, P.; Ratajczak, M.Z.; Zembala, M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol. Immunother. 2006, 55, 808–818. [Google Scholar] [CrossRef]
- Wang, Z.; von Au, A.; Schnölzer, M.; Hackert, T.; Zöller, M. CD44v6-competent tumor exosomes promote motility, invasion and cancer-initiating cell marker expression in pancreatic and colorectal cancer cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koistinen, V.; Härkönen, K.; Kärnä, R.; Arasu, U.T.; Oikari, S.; Rilla, K. EMT induced by EGF and wounding activates hyaluronan synthesis machinery and EV shedding in rat primary mesothelial cells. Matrix Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lenart, M.; Rutkowska-Zapala, M.; Baj-Krzyworzeka, M.; Szatanek, R.; W?glarczyk, K.; Smallie, T.; Ziegler-Heitbrock, L.; Zembala, M.; Siedlar, M. Hyaluronan carried by tumor-derived microvesicles induces IL-10 production in classical (CD14++CD16−) monocytes via PI3K/Akt/mTOR-dependent signalling pathway. Immunobiology 2017, 222. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; D’Ascenzo, S.; Borsotti, P.; Giavazzi, R.; Pavan, A.; Dolo, V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Dolo, V.; Ginestra, A.; Cassarà, D.; Violini, S.; Lucania, G.; Torrisi, M.R.; Nagase, H.; Canevari, S.; Pavan, A.; Vittorelli, M.L. Selective localization of matrix metalloproteinase 9, β 1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells. Cancer Res. 1998, 58, 4468–4474. [Google Scholar]
- McCarthy, J.B.; El-Ashry, D.; Turley, E.A. Hyaluronan, cancer-associated fibroblasts and the tumor microenvironment in malignant progression. Front. Cell Dev. Biol. 2018, 6, 48. [Google Scholar] [CrossRef]
- Szczepanik, A.; Sierzega, M.; Drabik, G.; Pituch-Noworolska, A.; Kołodziejczyk, P.; Zembala, M. CD44+ cytokeratin-positive tumor cells in blood and bone marrow are associated with poor prognosis of patients with gastric cancer. Gastric Cancer 2019. [Google Scholar] [CrossRef] [Green Version]
- Pituch-Noworolska, A.; Wiȩckiewicz, J.; Krzeszowiak, A.; Stachura, J.; Ruggiero, I.; Gawlicka, M.; Szczepanik, A.; Karcz, D.; Popiela, T.; Zembala, M. Evaluation of circulating tumour cells expressing CD44 variants in the blood of gastric cancer patients by flow cytometry. Anticancer Res. 1998, 18, 3747–3752. [Google Scholar]
- Tian, W.; Liu, S.; Li, B. Potential role of exosomes in cancer metastasis. BioMed Res. Int. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoraki, M.N.; Matsumoto, A.; Beccard, I.; Hoffmann, T.K.; Whiteside, T.L. CD44v3 protein-carrying tumor-derived exosomes in HNSCC patients’ plasma as potential noninvasive biomarkers of disease activity. Oncoimmunology 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Welton, J.L.; Khanna, S.; Giles, P.J.; Brennan, P.; Brewis, I.A.; Staffurth, J.; Mason, M.D.; Clayton, A. Proteomics analysis of bladder cancer exosomes. Mol. Cell. Proteom. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.S.; Choi, D.Y.; Hong, B.S.; Jang, S.C.; Kim, D.K.; Lee, J.; Kim, Y.K.; Kim, K.P.; Gho, Y.S. Quantitative proteomics of extracellular vesicles derived from human primary and metastatic colorectal cancer cells. J. Extracell. Vesicles 2012. [Google Scholar] [CrossRef]
- Purushothaman, A.; Bandari, S.K.; Chandrashekar, D.S.; Jones, R.J.; Lee, H.C.; Weber, D.M.; Orlowski, R.Z. Chondroitin sulfate proteoglycan serglycin influences protein cargo loading and functions of tumor-derived exosomes. Oncotarget 2017. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cheng, K.; Zhang, G.; Jia, Z.; Yu, Y.; Guo, J.; Hua, Y.; Guo, F.; Li, X.; Zou, W.; et al. Enrichment of CD44 in exosomes from breast cancer cells treated with doxorubicin promotes chemoresistance. Front. Oncol. 2020. [Google Scholar] [CrossRef]
- Kato, T.; Mizutani, K.; Kawakami, K.; Fujita, Y.; Ehara, H.; Ito, M. CD44v8-10 mRNA contained in serum exosomes as a diagnostic marker for docetaxel resistance in prostate cancer patients. Heliyon 2020. [Google Scholar] [CrossRef]
- Chiang, S.P.H.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [Green Version]
- Di Vizio, D.; Morello, M.; Dudley, A.C.; Schow, P.W.; Adam, R.M.; Morley, S.; Mulholland, D.; Rotinen, M.; Hager, M.H.; Insabato, L.; et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am. J. Pathol. 2012, 181. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 71. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal MIR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Klingbeil, P.; Marhaba, R.; Jung, T.; Kirmse, R.; Ludwig, T.; Zöller, M. CD44 variant isoforms promote metastasis formation by a tumor cell-matrix cross-talk that supports adhesion and apoptosis resistance. Mol. Cancer Res. 2009, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Gross, W.; Zöller, M. CD44v6 coordinates tumor matrix-triggered motility and apoptosis resistance. J. Biol. Chem. 2011, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34. [Google Scholar] [CrossRef]
- Yen, E.Y.; Miaw, S.C.; Yu, J.S.; Lai, I.R. Exosomal TGF-β1 is correlated with lymphatic metastasis of gastric cancers. Am. J. Cancer Res. 2017, 7. [Google Scholar]
- Sidhu, S.S.; Mengistab, A.T.; Tauscher, A.N.; LaVail, J.; Basbaum, C. The microvesicle as a vehicle for EMMPRin in tumor-stromal interactions. Oncogene 2004, 23, 956–963. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67. [Google Scholar] [CrossRef] [Green Version]
- Roh-Johnson, M.; Bravo-Cordero, J.J.; Patsialou, A.; Sharma, V.P.; Guo, P.; Liu, H.; Hodgson, L.; Condeelis, J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 2014, 33. [Google Scholar] [CrossRef] [Green Version]
- Courtneidge, S.A.; Azucena, E.F.; Pass, I.; Seals, D.F.; Tesfay, L. The SRC substrate Tks5, podosomes (Invadopodia), and cancer cell invasion. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2005; Volume 70. [Google Scholar]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-Induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [Green Version]
- Seals, D.F.; Azucena, E.F.; Pass, I.; Tesfay, L.; Gordon, R.; Woodrow, M.; Resau, J.H.; Courtneidge, S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 2005, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.M.C.; Chen, G.; Dayton, T.L.; Kim-Kiselak, C.; Hoersch, S.; Whittaker, C.A.; Bronson, R.T.; Beer, D.G.; Winslow, M.M.; Jacks, T. Differential Tks5 isoform expression contributes to metastatic invasion of lung adenocarcinoma. Genes Dev. 2013, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miletti-González, K.E.; Murphy, K.; Kumaran, M.N.; Ravindranath, A.K.; Wernyj, R.P.; Kaur, S.; Miles, G.D.; Lim, E.; Chan, R.; Chekmareva, M.; et al. Identification of function for CD44 intracytoplasmic domain (CD44-ICD): Modulation of matrix metalloproteinase 9 (MMP-9) transcription via novel promoter response element. J. Biol. Chem. 2012, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.-M.; Luo, D.-X.; Zhang, J. SDF-1/CXCR7 axis regulates the proliferation, invasion, adhesion, and angiogenesis of gastric cancer cells. World J. Surg. Oncol. 2016, 14, 256. [Google Scholar] [CrossRef] [Green Version]
- Mook, O.R.F.; Frederiks, W.M.; Van Noorden, C.J.F. The role of gelatinases in colorectal cancer progression and metastasis. Biochim. Biophys. Acta Rev. Cancer 2004, 1705, 69–89. [Google Scholar] [CrossRef]
- Mi, Z.; Bhattacharya, S.D.; Kim, V.M.; Guo, H.; Talbotq, L.J.; Kuo, P.C. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 2011, 32. [Google Scholar] [CrossRef]
- Ma, L.; Dong, L.; Chang, P. CD44v6 engages in colorectal cancer progression. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14. [Google Scholar] [CrossRef] [Green Version]
- Orian-Rousseau, V.; Sleeman, J. CD44 is a multidomain signaling platform that integrates extracellular matrix cues with growth factor and cytokine signals. Adv. Cancer Res. 2014, 123, 231–254. [Google Scholar] [PubMed]
- Strell, C.; Entschladen, F. Cell communication and signaling extravasation of leukocytes in comparison to tumor cells. Cell Commun. Signal. 2008, 13, 1–13. [Google Scholar]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinkovich, A.; Tavor, S.; Avigdor, A.; Kahn, J.; Brill, A.; Petit, I.; Goichberg, P.; Tesio, M.; Netzer, N.; Naparstek, E.; et al. Functional CXCR4-expressing microparticles and SDF-1 correlate with circulating acute myelogenous leukemia cells. Cancer Res. 2006, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Guo, J.; Yang, M.; Zhu, X.; Cao, X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J. Immunol. 2011, 186. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.J.; Hoos, A.; Bauer, J.; Boettcher, S.; Knust, M.; Weber, A.; Simonavicius, N.; Schneider, C.; Lang, M.; Stürzl, M.; et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 2012, 22. [Google Scholar] [CrossRef] [Green Version]
- Braumüller, H.; Gansauge, S.; Ramadani, M.; Gansauge, F. CD44v6 cell surface expression is a common feature of macrophages and macrophage-like cells—Implication for a natural macrophage extravasation mechanism mimicked by tumor cells. FEBS Lett. 2000, 476. [Google Scholar] [CrossRef] [Green Version]
- Abecasis, M.; Cross, N.C.P.; Brito, M.; Ferreira, I.; Sakamoto, K.M.; Hijiya, N.; Score, J.; Gale, R.P. Is cancer latency an outdated concept? Lessons from chronic myeloid leukemia. Leukemia 2020, 34. [Google Scholar] [CrossRef]
- Leung, E.L.H.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.C.; Cheng, L.C.; Sihoe, A.D.L.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Hoe, S.L.L.; Tan, L.P.; Abdul Aziz, N.; Liew, K.; Teow, S.Y.; Abdul Razak, F.R.; Chin, Y.M.; Mohamed Shahrehan, N.A.; Chu, T.L.; Mohd Kornain, N.K.; et al. CD24, CD44 and EpCAM enrich for tumour-initiating cells in a newly established patient-derived xenograft of nasopharyngeal carcinoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Lane, R.; Simon, T.; Vintu, M.; Solkin, B.; Koch, B.; Stewart, N.; Benstead-Hume, G.; Pearl, F.M.G.; Critchley, G.; Stebbing, J.; et al. Cell-derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun. Biol. 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Xu, Y.; Gu, P.; Wang, W.; Li, J.; Ge, Y.; Wang, H. Exosomal CD44 cooperates with integrin α6β4 to support organotropic metastasis via regulating tumor cell motility and target host cell activation. Engineering 2020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | Outcome | Reference | |
|---|---|---|---|
| CD44s | ERM/MAPK | Cell division, proliferation | [50] |
| PI3K/AKT/NFκB | Inflammatory cytokine release | [51] | |
| Migration of ICD fragment into the nucleus | Metastasis, survival | [52] | |
| RHAMM/cSrc/RAS | Migration | [53] | |
| CD44v | ERM/FAK/Paxillin | Angiogenesis | [54] |
| Src/AKT/HIF1α | Metabolic shift (glycolysis increase) | [49] | |
| Met/PI3K/AKT | Cell invasion | [55] | |
| Met/Snail/MMP | Cell division, proliferation | [56] |
| Type of Cancer | CD44 form Carried by TEVs | Cargo of TEVs | Role | Reference |
|---|---|---|---|---|
| Head and neck | CD44s, CD44v3 | PD-L1, FasL, TGF-β, EGFR | Immunosupressive biomarker of disease | [82] |
| Bladder | CD44s | MUC-1, EMMPRIN, HLA I, integrins | disease monitoring, | [83] |
| Colon | CD44s | HA, EGFR, ADAM10 | Cancer progression | [42,84,85] |
| CD44v6 | MMP2,-7,-9,-13,-14,-17, ADAM10, ADAM17, CD26, uPAR TIMP1, TIMP 2 CXCR4, MET, EpCAM, Tspan8, integrins | Motility and invasion promotion | [71] | |
| Pancreas | CD44s, CD44v6 | MET, EpCAM, integrin, CXCR4, uPAR, ADAM17, MMP2, 9, 7, 13, 14 | Motility and invasion promotion | [71] |
| CD44s, CD44v7–8 | EpCAM, EMMPRIN, mRNA for VEGF, IL-8, HGF | interaction with monocytes | [70] | |
| Ovarian | CD44s | n.a. | Invasion promotion | [69] |
| Breast | CD44s | n.a. | Drug resistance | [86] |
| Gastric | CD44s | HA | ECM interactions | [6] |
| Prostate | CD44s, CD44v8–10 | CD44v8–10 mRNA | Drug resistance | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szatanek, R.; Baj-Krzyworzeka, M. CD44 and Tumor-Derived Extracellular Vesicles (TEVs). Possible Gateway to Cancer Metastasis. Int. J. Mol. Sci. 2021, 22, 1463. https://doi.org/10.3390/ijms22031463
Szatanek R, Baj-Krzyworzeka M. CD44 and Tumor-Derived Extracellular Vesicles (TEVs). Possible Gateway to Cancer Metastasis. International Journal of Molecular Sciences. 2021; 22(3):1463. https://doi.org/10.3390/ijms22031463
Chicago/Turabian StyleSzatanek, Rafał, and Monika Baj-Krzyworzeka. 2021. "CD44 and Tumor-Derived Extracellular Vesicles (TEVs). Possible Gateway to Cancer Metastasis" International Journal of Molecular Sciences 22, no. 3: 1463. https://doi.org/10.3390/ijms22031463