Abstract
Human adenoviruses (HAdVs) display a wide range of tissue tropism and can cause an array of symptoms from mild respiratory illnesses to disseminated and life-threatening infections in immunocompromised individuals. However, no antiviral drug has been approved specifically for the treatment of HAdV infections. Herein, we report our continued efforts to optimize salicylamide derivatives and discover compound 16 (JMX0493) as a potent inhibitor of HAdV infection. Compound 16 displays submicromolar IC50 values, a higher selectivity index (SI > 100) and 2.5-fold virus yield reduction compared to our hit compound niclosamide. Moreover, unlike niclosamide, our mechanistic studies suggest that the antiviral activity of compound 16 against HAdV is achieved through the inhibition of viral particle escape from the endosome, which bars subsequent uncoating and the presentation of lytic protein VI.
1. Introduction
Human adenoviruses (HAdVs) are common pathogens with broad tissue tropism and are frequently responsible for infections displaying mild to moderate respiratory and gastrointestinal symptoms as well as conjunctivitis, notably in pediatric populations and among active duty military service members. HAdVs are characterized by a linear double-stranded DNA genome, ranging from 34–36 kb in size, surrounded by a nonenveloped, icosahedral capsid [1]. Presently, HAdVs compromise more than 100 classified serotypes distributed among seven species (HAdV A–G), which display a large variance in infection and symptom severity [2,3]. In healthy individuals with normal immune function, HAdV infections are often self-limiting and are rarely associated with severe disease. However, in immunocompromised individuals and other at-risk groups, such as solid-organ transplant (SOT) and allogenic hematopoietic stem cell transplant (allo-HSCT) recipients, HAdV infections can result in life-threatening illness and infection-related medical complications [4,5]. Pediatric allo-HSCT recipients are especially high-risk, exemplified by a frequency of infections recorded as high as 42–47% with the mortality rate up to 80% in cases with disseminated disease [4,6,7]. Additionally, following increasingly accurate molecular diagnostic technology development, HAdV infections have been found to be involved in both isolated cases and outbreaks of community-acquired pneumonia (CAP) in otherwise healthy individuals [8,9,10,11,12,13].
Despite the consequences of acquiring disseminated HAdV infection, there are no antiviral drugs specifically approved for the treatment of HAdV infections [14,15,16,17,18,19,20,21]. Currently, intravenous cidofovir (Figure 1) is used off-label to treat HAdV-associated diseases in many transplant clinics. Cidofovir, an acyclic nucleotide phosphonate cytosine analogue, is considered a broad-spectrum antiviral agent against DNA viruses and has shown efficacy against all HAdV serotypes in vitro, acting as a chain terminator during DNA replication [22]. However, cidofovir lacks sufficient oral bioavailability and is highly subject to renal elimination; thus, even its intravenous administration can result in notable nephrotoxicity and myelotoxicity, necessitating the routine coadministration of probenecid to mitigate nephrotoxicity [14]. In efforts to utilize the efficacy against HAdVs, a series of lipid ester derivatives of cidofovir were synthesized that displayed meaningfully decreased nephrotoxicity by reducing kidney exposure to the drug, and were also marked by increased oral bioavailability and cellular uptake due to the lip moiety, which is cleaved inside the cell by phospholipase to transform the prodrug into cidofovir [23,24,25]. Among these derivatives, the alkoxyalkyl ester prodrug brincidofovir (BCV, CMX001, 3-hexadecyloxy-1-propanol-cidofovir, 2) showed promising results in phase II and III clinical trials (NCT01231344 and NCT02087306), which heightened interest in its potential to be developed as the first approved treatment for HAdV infections [26,27,28,29]. Unfortunately, long-term trials of BCV treatment led to adverse effects such as diarrhea in some patients and also highlighted that alternative administration strategies should be considered [29]. Therefore, there is still an urgent need to develop highly effective antivirals against HAdV infections that are characterized by a lack of toxicity, reduced adverse effects, and increased oral bioavailability.
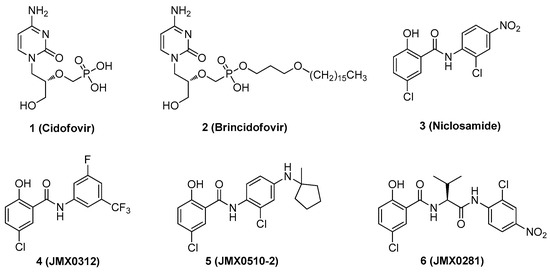
Figure 1.
Representative human adenovirus (HAdV) inhibitors.
We discovered that niclosamide (3), an FDA-approved anthelmintic drug that is a part of our ongoing antiviral drug discovery and development program [30,31,32,33,34,35,36], significantly inhibited HAdV infection with an IC50 value of 0.6 μM, displayed moderate cytotoxicity (CC50 = 22.9 μM), and was characterized as having a narrow safety window, as shown by a selectivity index (SI) of 38.2 [37]. To improve potency and decrease toxicity, we focused our previous strategies for chemical modification on the aniline moiety substituents and changes to the amide linker, whereby a series of salicylamide derivatives were discovered as potent anti-HAdV inhibitors with improved safety windows [38,39]. Among these derivatives, the 3′-fluoro-5′-trifluoromethyl substituted compound 4 and the 2′-chloro-5′-(1-methylcyclopentyl)amino substituted compound 5 showed improvements in potency (IC50 = 0.18 μM and 0.27 μM, respectively), decreased cytotoxicity (CC50 = 120.0 μM and 156.8 μM, respectively) and dramatically increased the selectivity index (SI = 666.7 and 580.7, respectively), whereas compound 6 modified with a L-valine linker maintained the same level of potency (IC50 = 0.45 μM) with a decreased cytotoxicity (CC50 = 200.0 μM) and increased selectivity index (SI = 444.4), compared to the hit compound niclosamide. However, all of these lead compounds yielded from our previous optimization efforts shared the similarity by containing an aniline group to form the amide scaffolds like niclosamide, and whether this moiety can be altered with more conformationally flexible alkyl groups remains unclear. Herein, as depicted in Figure 2, we further explored the structure–activity relationship (SAR) studies by replacing the aniline moiety of niclosamide with various alkylamine groups to identify new antiviral agents with potential action mechanisms distinct from that of niclosamide. This work highlights the discovery of a unique compound 16 (JMX0493), which displays an improved selectivity index while acting as a potent inhibitor of HAdV infection by targeting the HAdV entry pathway that prevents viral particle disassembly and subsequent release from the endosome.
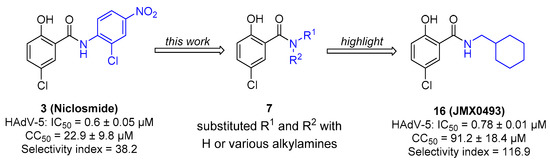
Figure 2.
The drug design for this work, which highlights the discovery of new compound 16 (JMX0493) with both improved selectivity index and capability of inhibiting the escape from the endosome of the viral particles.
2. Results and Discussion
2.1. Chemistry
The general synthesis of salicylamide derivatives is shown in Scheme 1. Direct substitution of methyl 5-chloro-2-hydroxybenzoate with a variety of amines in methanol afforded derivatives 11–15, 18–25, and 28. As shown in Scheme 2, condensation of 5-chloro-2-hydroxybenzoic acid with cyclohexylmethanamine or (S)-1-cyclohexylethanamine provided compounds 16 and 17, respectively. 5-Chloro-2-methoxybenzoic acid was coupled with piperidine or 1-acetylpiperazine followed by demethylation with BBr3 to give compounds 26 and 27, respectively. Efficient and accessible synthetic methodology is imperative in antiviral discovery efforts and remains a consideration in our molecular design along with robust SAR studies discussed in the following section.
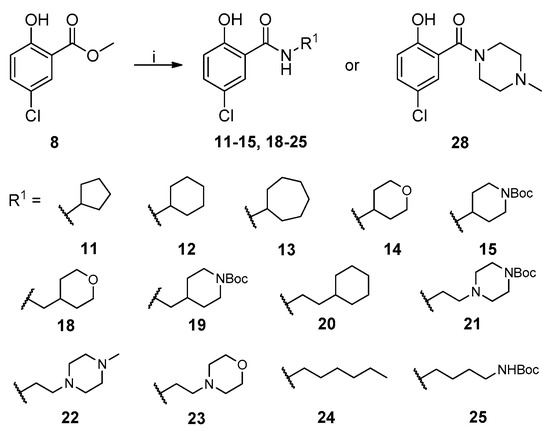
Scheme 1.
Synthetic route to achieve compounds 11–15, 18–25, and 28. Reagents and conditions: (i) R1NH2 or 1-methylpiperazine, CH3OH, r.t. to 80 °C, 48–96 h.
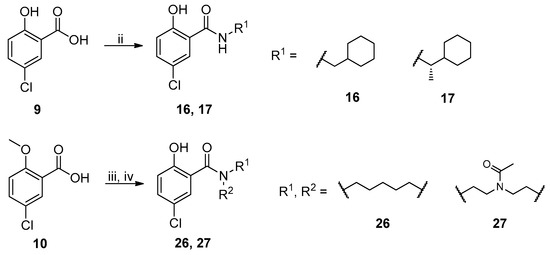
Scheme 2.
Synthetic route to achieve compounds 16, 17, 26, and 27. Reagents and conditions: (ii) R1NH2, EDCI, DMAP, DCM, r.t., 12 h. (iii) piperidine or 1-acetylpiperazine, EDCI, DMAP, DCM, r.t., 2 h; (iv) BBr3, DCM, −78 °C to 0 °C, 12 h.
2.2. In Vitro Evaluation of Human Adenovirus Inhibition
The newly synthesized compounds were first screened in the plaque assay, measuring the inhibition of HAdV plaque formation. As shown in Table 1, we first substituted the aniline moiety of niclosamide with simple cycloalkanamines (11–15). When we increased the ring size of the cycloalkyl groups, the corresponding compounds 11–13 showed a trend where ring size correlated with increased potency with inhibition values of 22.6%, 57.6%, and 99.6% at 10 μM, respectively. Compound 13 with the N-cycloheptyl substitution displayed micromolar potency (IC50 = 4.7 μM) and similar cytotoxicity (CC50 = 30.9 μM) to niclosamide. Interestingly, the tetrahydropyran derivative 14 was completely inactive at 10 μM, whereas the N-Boc-piperidine-substituted compound 15 inhibited HAdV plaque formation with a percentage of 52.2%. Inserting one carbon atom as a linker between the amide and cycloalkyl produced compounds 16–19. Excitingly, the N-cyclohexylmethyl substituted derivative 16 exhibited submicromolar potency (IC50 = 0.78 μM) and, in contrast to niclosamide, displayed significantly decreased cytotoxicity (CC50 = 91.2 μM) and an increased selectivity index (SI = 116.9). The introduction of an (S)-methyl on the methylene linker (17) retained the same level of potency (IC50 = 0.51 μM) but significantly increased the cytotoxicity (CC50 = 23.0 μM) as compared with derivative 16, steering our efforts away from similar such modifications. Compound 18 with a tetrahydropyran moiety showed no anti-HAdV activity, whereas the N-Boc protected derivative 19 was active with an inhibition percent of 88.5% against HAdV plaque formation at 10 μM. By adding an additional carbon atom in the linker to achieve the N-cyclohexylethyl derivative 20, we observed a slight loss of potency (IC50 = 1.0 μM) but also diminished cytotoxicity (CC50 = 66.3 μM) and improved selectivity index (SI = 64.3) compared to niclosamide. However, building on the two-carbon linker theme, we found that neither piperazine analogue (21 and 22) nor the morpholine analogue 23 showed meaningful inhibitory activity against HAdV plaque formation up to 10 μM. The lack of inhibitory activity by 23 and 14 may be associated with limited tolerance for electronegative atoms within R2 position substituents and is under subsequent investigation. Surprisingly, derivative 24 with a linear hexylamine substitution maintained single digit micromolar potency with an IC50 value of 3.73 μM. As expected, N-Boc protected derivative 25 was also active against HAdV with inhibition of 74.0% at 10 μM. To probe molecular space constraints, secondary amine intramolecular ring moieties were applied but found not to be tolerated in any configuration tested, as shown by the activity data for compounds 26–28. Based on our previous work and the SAR reported herein, we hypothesize that the target binding site accommodating the R2 moiety is preferentially engaged by simple hydrophobic moieties that are either of moderate length (e.g., one carbon linker) or shortened, bulky groups (e.g., N-cycloheptyl moiety), and are devoid of terminal electronegative atoms as seen in tetrahydropyran and morpholine derivatives. Larger, more complex moieties may be tolerated, but at a cost of incorporating additional functional groups that are metabolically labile or contributors to cytotoxicity. Thus, compound 16 was of keen interest due to the conservative N-cyclohexylmethyl substitution and its noteworthy inhibition activity, low cytotoxicity, and high selectivity index (SI > 100).

Table 1.
Inhibition of HAdV in plaque assay, cytotoxicity, and selectivity index for compounds 11–28.
We next evaluated the anti-HAdV effect of compound 16 using a virus burst assay that can measure the ability of a pharmacological agent to suppress the production of new viral particles. As shown in Table 2, the addition of compound 16 resulted in a 208-fold overall reduction in virus yield, 2.5 times greater than the reduction observed by niclosamide addition.
Table 2.
Virus yield reduction for derivative 16.
2.3. Antiviral Mechanism of Action Studies
The HAdV replicative cycle is a multistage process characterized by numerous, low-redundancy steps that may be targeted to inhibit viral attachment, entry, replication, and/or escape. Thus, we investigated several key points in its replicative cycle to identify a potential target responsible for the antiviral activity of compound 16.
2.3.1. Impact of Compound 16 on HAdV Replication
We first examined the effect that compound 16 had on the HAdV DNA replication process. As viral replication is dependent upon efficient DNA replication, we performed quantitative real-time PCR to measure HAdV DNA replication in the presence of compound 16. After one round of infection, DNA was extracted at 24 h, avoiding the influence of newly generated viral particles derived from subsequent rounds of infection occurring at 32–36 h timepoints [19]. As shown in Figure 3, compound 16 displayed significant activity in the qPCR assay, inhibiting or delaying HAdV-5 DNA replication by 50% at a concentration of 7.8 μM (p ≤ 0.001) when compared to the DMSO treated control, whereas niclosamide prevented any detectable DNA replication. This result indicates that compound 16 likely targets, at least partially, the HAdV DNA replication process. The decrease in HAdV DNA copy number in the presence of compound 16 suggests that this molecule could be interfering directly with proteins involved in HAdV DNA replication or with an earlier step in its replicative cycle, through which interference would indirectly downregulate DNA replication.
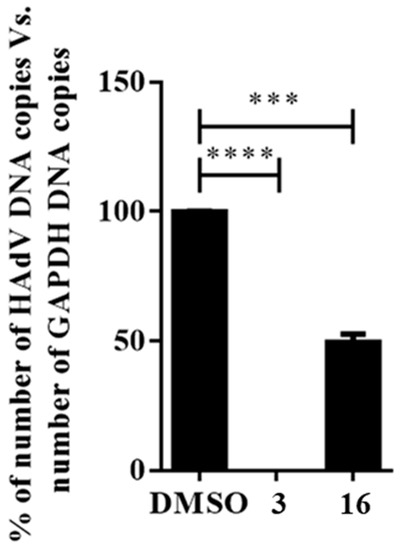
Figure 3.
Effect of compound 16 on the HAdV DNA replication. Concentration of niclosamide (3) was 5 μM and 10-fold IC50 concentration obtained in the plaque assay for compound 16. Bars represent means ± SD of triplicate samples. *** p ≤ 0.001, **** p ≤ 0.0001.
2.3.2. Time of Addition Assay
To obtain a more complete picture and pinpoint the potential step in the HAdV replicative cycle targeted by compound 16, we measured the time dependence of compound addition on its ability to inhibit HAdV. HAdV has been shown to be enter the cell within 5 min of viral attachment, to escape the endosome after 15 min, and to attach to the nuclear pore complex after 35−45 min postinfection (p.i.). Our results showed that the antiviral activity of compound 16 exhibited a time-dependent decrease in activity Figure 4, especially apparent at early time points where we observed a decrease in its inhibitory activity from 95.3% inhibition at 0 min p.i. to 30.7% inhibition at 20 min p.i. Collectively, these results indicate that compound 16 inhibits an early step in the virus cycle, after cell attachment and before entry into the cell nucleus.
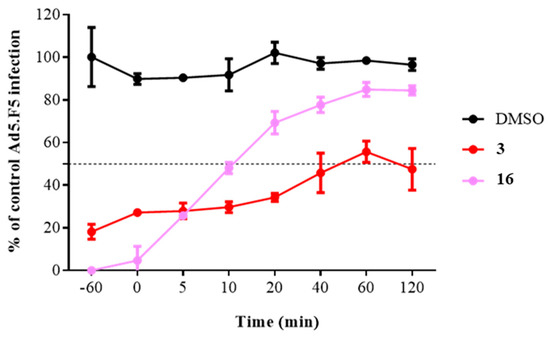
Figure 4.
Effect of compound 16 on HAdV infection at different time points. The concentrations used were 5 μM for niclosamide (3) and 10-fold IC50 concentration obtained in the plaque assay for analog 16. Line charts represent means ± SD of duplicate samples.
To confirm if the antiviral activity of compound 16 was dependent on a step between HAdV entry and the arrival to the nucleus, we performed an assay to quantify the HAdV genome accessibility to the nucleus. Once HAdV escapes from the endosome, the partially uncoated capsid is transported to the cell nucleus, where further disassembly occurs and the HAdV genome is released into the cell nucleus for early gene transcription and subsequent replication. Thus, any inhibitory effect on that process will be reflected by the number of HAdV genomes that reach the host nucleus after a synchronized infection. Briefly, 45 min after infection, cells were recovered to separate the cytoplasmic and nuclear fractions. Then, HAdV DNA was isolated from each fraction and quantify by the real-time PCR. As shown in Figure 5, compound 16 inhibited more than 99% of the HAdV DNA accessibility to the nucleus and showed a higher inhibitory effect compared to the DMSO control experiment and niclosamide at 45 min postinfection (p ≤ 0.0001). At this point, our mechanistic approaches confirmed that the antiviral activity of compound 16 was targeting a step before HAdV transcription of early genes and DNA replication. Going backwards in the HAdV replicative cycle, the next process is to identify the precise step targeted by compound 16 through evaluating the HAdV escape from the endosome.
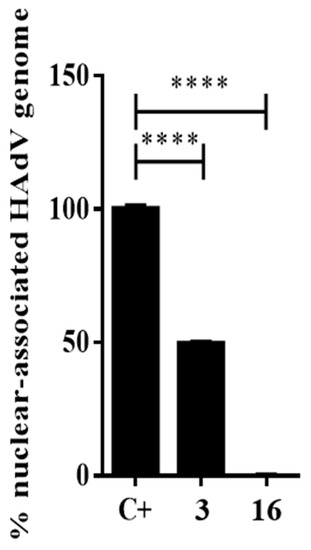
Figure 5.
Percentage of nuclear-associated HAdV genome of compound 16. Concentration of niclosamide (3) was 5 μM and 10-fold IC50 concentration obtained in the plaque assay for compound 16. Bars represent means ± SD of triplicate samples. **** p ≤ 0.0001.
2.3.3. Impacts of Compound 16 on HAdV Escape from the Endosome
Upon attachment to its cellular receptors, HAdV particles are internalized by endocytosis into the cells and viral particles undergo partial disassembly inside the early endosomes, resulting in the release of protein VI from the interior of the capsid, which plays a key role in HAdV escape from the endosome [40]. To evaluate the potential influence of compound 16 in preventing the HAdV escape from the endosome, we used the α-sarcin co-delivery assay as a measurement of the ability of this compound to interfere with virus-mediated endosome lysis. Since the α-sarcin alone is unable to penetrate the cell, in this assay, the successful virus-mediated lysis of the endosome would result in ribotoxin-mediated inhibition of cellular protein synthesis. Increasing concentrations of HAdV in the presence or absence of compound 16 were mixed with α-sarcin and incubated with A549 cells. After metabolic labeling with methionine L-homopropargylglycine (HPG), cell samples were assayed for HPG incorporation into cellular proteins. As shown in Figure 6, cells treated with either the vehicle (DMSO) or niclosamide showed rates of HPG incorporation under 50% compared to a control without α-sarcin, independent of the virus concentration used. Interestingly, compound 16 showed a similar behavior as the entry-defective ts1 mutant HAdV that contains a mutation in the protease gene and fails to penetrate cell endosomes [41]. Compound 16 prevented HAdV-mediated endosome lysis at concentrations of the virus below 12 ng, similar to the effects observed by the ts1 mutant. This result, together with our time of addition assay, showing a decrease of the inhibitory effect after 15–20 min p.i, suggests that the mechanism for inhibition of this compound is likely related to the blockage of HAdV escape from the endosome.
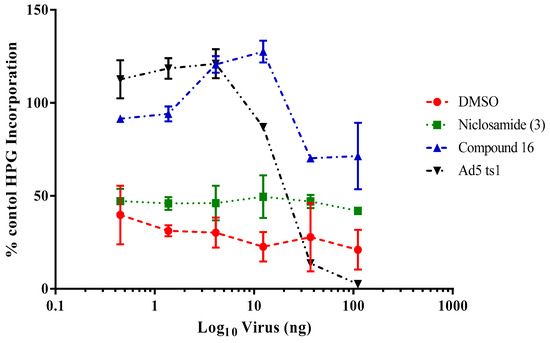
Figure 6.
Activity of compound 16 on HAdV-mediated endosomolysis in a α-sarcin assay. The concentrations used were 5 μM for niclosamide (3) and 10-fold IC50 concentration obtained in the plaque assay for derivative 16. Results represent means ± SD of triplicate assays.
The studies described above delineate the step of infection that is targeted by compound 16; however, the precise molecular mechanism involved in its antiviral activity remained to be established. One alternative mode of action was that compound 16 could block HAdV-mediated lysis of the endosome by stabilizing the virus capsid, thereby preventing uncoating. Indeed, human defensins and neutralizing antibodies have been previously shown to act through this mechanism [42]. To determine whether compound 16 impacts HAdV uncoating, we used a thermostability assay that mimics virus disassembly in the endosome, which was previously described by Wiethoff et al. [40], with a few modifications. In this assay, temperatures above 48 °C promote selective removal of the virus vertex region. HAdV-5 was incubated with or without 50 µM concentrations of compound 16 at temperatures from 37 °C to 52 °C and then added to cells to evaluate the viability of the viruses. Viruses incubated with either compound 16 or DMSO at 37 °C, 40 °C, and 44.5 °C were largely intact, showing similar rates of infection measured by the number of cells expressing GFP Figure 7. Upon heating HAdV-5 to 48 °C or above, in those nontreated with compound 16, there was no GFP expression 24 h postinfection. However, in wells with the addition of 16, significant levels of GFP expression were observed for those viruses heated to 48 °C, and some residual expression remained at 52 °C. These findings suggest that compound 16 may prevent HAdV endosomal escape by stabilizing the viral capsid and subsequently inhibiting endosomal membrane lysis [43].
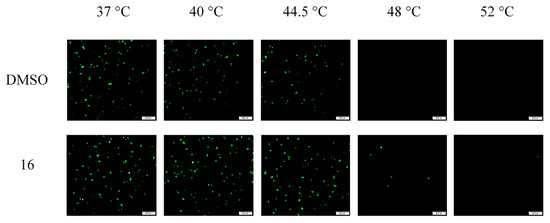
Figure 7.
Compound 16 stabilizes HAdV capsid. The concentration used was 50 μM. Data are representative of three experiments.
2.4. Synergistic Activity of the Selected Compounds
In previous reports [38,39], we found three compounds—4 (JMX0312), 5 (JMX0510-2), and 6 (JMX0281)—with high activity against HAdV, targeting different steps in the HAdV replicative cycle. We hypothesize that the double and triple combination of these compounds with compound 16 (JMX0493) should significantly increase their antiviral activity. To evaluate our hypothesis, we conducted a combination study based on the Chou–Talalay method for drug combination using the CalcuSyn software [44]. The constant ratio for each combination was selected based on the IC50 values for each drug. The data for all the combinations showed good conformity with the mass action law principle (r = 0.87–0.95) Table 3. Two out of three double combinations showed a synergistic effect, and concretely, the combination of compounds 4 and 16 (ratio 1:4.3) showed strong synergism (ED50 = 0.26) and the combination of compounds 6 and 16 (ratio 1:1.73) was also classified as synergistic (ED50 = 0.40), while the combination of compounds 5 and 16 (ratio 1:2.9) showed only an additive effect (ED50 = 1.09). For the triple combinations, one of them (the combination of compounds 4, 5, and 16; ratio 1:1.5:4.3) exhibited a synergistic effect with an ED50 value of 0.33, while the other triple combination (compounds 5, 6, and 16; ratio 1:1.7:2.9) showed a clear antagonistic effect (ED50 = 2.01), probably due to compound interactions. It is worth noting that in our previous report, the combinatory index also increased when compound 5 was added to the mix, reaching additive or antagonistic values.
Table 3.
IC50 values in a plaque assay for each derivative and its combination. Combination index value was calculated with CalcuSyn software.
2.5. Impact of Compound 16 on HCMV Replication
The broad antiviral activity of compound 16 was tested using human cytomegalovirus (HCMV) in a DNA replication assay. To avoid the influence of newly generated viral particles from subsequent rounds of infection, HCMV DNA was extracted from HFF cells at 72 h postinfection and quantified in a single round of infection as a measurement of the DNA replication efficiency. The addition of compound 16 displayed significant reductions (p ≤ 0.001) in the quantification of total HCMV DNA, showing a 95% decrease compared to the DMSO control, as depicted in Figure 8.
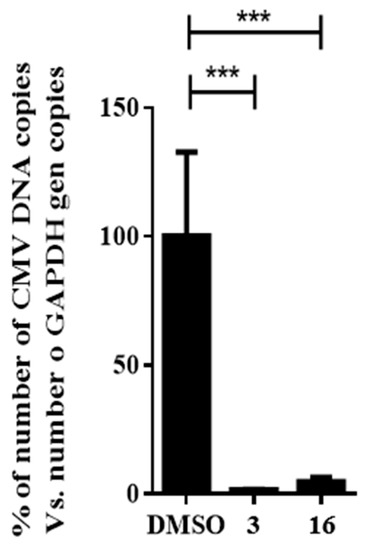
Figure 8.
Effect of compound 16 on HCMV replication. Concentration of niclosamide (3) was 5 μM and compound 16 was tested at 10-fold IC50 concentration obtained in the HAdV plaque assay. Bars represent means ± SD of triplicate samples. *** p ≤ 0.001.
3. Conclusions
In summary, salicylamide derivatives were designed, synthesized, and biologically evaluated for their antiviral activity against HAdV. Among these new molecules, compound 16 (JMX0493) maintained submicromolar potency against HAdV while displaying a markedly higher selectivity index (SI > 100) compared to the hit compound niclosamide. In addition, compound 16 was able to reduce virus yield to a 2.5-fold greater extent than niclosamide, indicative of a safer and more effective profile in virus yield reduction. Moreover, compound 16 demonstrated potential as a broad antiviral agent by showing a significant inhibition of HCMV DNA replication. As for its mechanism of action, unlike niclosamide, compound 16 showed a specific inhibition of HAdV infection via blocking the escape of viral particles from the endosome by stabilizing the viral capsid and preventing HAdV uncoating, thereby blocking subsequent viral migration to the nuclear membrane.
4. Materials and Methods
4.1. Chemistry
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Brucker-300 (1H, 600 and 300 MHz; 13C, 150 and 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. 1H and 13C NMR spectra of representative compounds are shown in Supplemental Figures (page S2–S19 in Supporting Information). Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained using a Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: nano-ESI spray voltage was 1.8 kV; capillary temperature was 275 °C and the resolution was 60,000; ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal Digital Melting Point Apparatus and were uncorrected. Purities of final compounds were established using analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (0.1% TFA was added into both acetonitrile and water). All biologically evaluated compounds are >95% pure.
General procedure A. Methyl 5-chloro-2-hydroxybenzoate (1.0 eq) was dissolved in methanol (10 mL/0.5 mmol) followed by addition of different amine (3.0 eq). The resulting mixture was stirred at r.t. ~80 °C for 48~96 h, and then concentrated. The residue was purified by preparative TLC to afford the final amide products.
5-Chloro-N-cyclopentyl-2-hydroxybenzamide (11). Compound 11 (116 mg, 90%) was prepared as a beige solid according to general procedure A (60 °C, 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and cyclopentylamine. HPLC purity 99.9% (tR = 18.07 min). 1H NMR (300 MHz, CDCl3) δ 12.34 (s, 1H), 7.37–7.26 (m, 2H), 6.90 (d, J = 8.7 Hz, 1H), 6.39 (d, J = 4.2 Hz, 1H), 4.45–4.27 (m, 1H), 2.21–1.98 (m, 2H), 1.83–1.45 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 168.6, 160.0, 133.9, 125.2, 123.3, 120.1, 115.5, 51.8, 33.1 (2C), 23.9 (2C). HRMS (ESI) calculated for C12H15ClNO2, 240.0791 (M + H)+; found, 240.0785.
5-Chloro-N-cyclohexyl-2-hydroxybenzamide (12). Compound 12 (71 mg, 51%) was prepared as a beige solid according to general procedure A (60 °C, 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and cyclohexylamine. HPLC purity 99.9% (tR = 18.85 min). 1H NMR (300 MHz, CDCl3) δ 12.36 (s, 1H), 7.35–7.27 (m, 2H), 6.90 (d, J = 8.4 Hz, 1H), 6.21 (d, J = 5.4 Hz, 1H), 4.00–3.86 (m, 1H), 2.06–1.94 (m, 2H), 1.83–1.60 (m, 3H), 1.48–1.13 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 168.1, 160.2, 134.0, 125.1, 123.3, 120.2, 115.6, 49.0, 33.0 (2C), 25.5, 25.0 (2C). HRMS (ESI) calculated for C13H17ClNO2, 254.0948 (M + H)+; found, 254.0941.
5-Chloro-N-cycloheptyl-2-hydroxybenzamide (13). Compound 13 (75 mg, 52%) was prepared as an off-white solid according to general procedure A (60 °C, 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and cycloheptylamine. HPLC purity 99.5% (tR = 19.58 min). 1H NMR (300 MHz, CDCl3) δ 12.36 (s, 1H), 7.34–7.27 (m, 2H), 6.90 (d, J = 9.0 Hz, 1H), 6.27 (d, J = 5.7 Hz, 1H), 4.20–4.03 (m, 1H), 2.08–1.93 (m, 2H), 1.74–1.47 (m, 10H). 13C NMR (75 MHz, CDCl3) δ 167.8, 160.2, 133.9, 125.1, 123.3, 120.2, 115.6, 51.2, 35.1 (2C), 28.0 (2C), 24.2 (2C). HRMS (ESI) calculated for C14H19ClNO2, 268.1104 (M + H)+; found, 268.1097.
5-Chloro-2-hydroxy-N-(tetrahydro-2H-pyran-4-yl)benzamide (14). Compound 14 (30 mg, 22%) was prepared as a light-yellow solid according to general procedure A (80 °C, 96 h), starting from methyl 5-chloro-2-hydroxybenzoate and 4-aminotetrahydropyran. HPLC purity 99.9% (tR = 15.48 min). 1H NMR (300 MHz, CDCl3) δ 12.18 (s, 1H), 7.38–7.29 (m, 2H), 6.98–6.88 (m, 1H), 6.21 (d, J = 6.6 Hz, 1H), 4.26–4.12 (m, 1H), 4.07–3.96 (m, 2H), 3.59–3.46 (m, 2H), 2.05–1.91 (m, 2H), 1.69–1.52 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 168.4, 160.3, 134.3, 125.1, 123.4, 120.3, 115.3, 66.8 (2C), 46.5, 33.1 (2C). HRMS (ESI) calculated for C12H15ClNO3, 256.0740 (M + H)+; found, 256.0734.
tert-Butyl 4-(5-chloro-2-hydroxybenzamido)piperidine-1-carboxylate (15). Compound 15 (24 mg, 13%) was prepared as an off-white solid according to general procedure A (80 °C, 96 h), starting from methyl 5-chloro-2-hydroxybenzoate and 4-aminopiperidine-1-carboxylic acid tert-butyl ester. HPLC purity 98.8% (tR = 18.51 min). 1H NMR (300 MHz, CDCl3) δ 12.21 (s, 1H), 7.43–7.23 (m, 2H), 6.96–6.88 (m, 1H), 6.44 (d, J = 6.6 Hz, 1H), 4.20–4.02 (m, 3H), 2.97–2.80 (m, 2H), 2.05–1.94 (m, 2H), 1.52–1.40 (m, 11H). 13C NMR (75 MHz, CDCl3) δ 168.5, 160.3, 154.9, 134.2, 125.3, 123.4, 120.3, 115.4, 80.1, 47.5, 42.9 (2C), 32.0 (2C), 28.6 (3C). HRMS (ESI) calculated for C17H24ClN2O4, 355.1425 (M + H)+; found, 355.1417.
5-Chloro-N-(cyclohexylmethyl)-2-hydroxybenzamide (16). To a solution of cyclohexanemethylamine (200 mg, 1.77 mmol), 5-chlorosalicylic acid (244 mg, 1.41 mmol) and DMAP (18 mg, 0.14 mmol) in 20 mL of DCM were added EDCI (509 mg, 2.66 mmol) at 0 °C. The resulting mixture was stirred at r.t. for 24 h and then concentrated. The residue was purified by column chromatography (Hex/EtOAc = 10/1) to afford compound 16 (240 mg, 63%) as a light-yellow solid. HPLC purity 99.9% (tR = 19.68 min). 1H NMR (300 MHz, CDCl3) δ 12.22 (brs, 1H), 7.36–7.28 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.43 (s, 1H), 3.28 (t, J = 6.3 Hz, 2H), 1.82–1.50 (m, 6H), 1.34–1.10 (m, 3H), 1.06–0.90 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.0, 160.2, 134.0, 125.0, 123.4, 120.2, 115.5, 46.1, 38.0, 31.0 (2C), 26.4, 25.9 (2C). HRMS (ESI) calculated for C14H19ClNO2, 268.1104 (M + H)+; found, 268.1101.
(S)-5-Chloro-N-(1-cyclohexylethyl)-2-hydroxybenzamide (17). Compound 17 was prepared using a procedure similar to that used to prepare compound 16 starting from 5-chlorosalicylic acid and (S)-1-cyclohexylethanamine. The title compound was obtained (138 mg, 39%) as an off-white solid. HPLC purity 99.6% (tR = 20.25 min). 1H NMR (300 MHz, CDCl3) δ 12.36 (s, 1H), 7.33 (dd, J = 8.7, 2.4 Hz, 1H), 7.29 (d, J = 2.4 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H), 6.03 (d, J = 7.5 Hz, 1H), 4.12–3.98 (m, 1H), 1.85–1.61 (m, 5H), 1.50–1.38 (m, 1H), 1.32–0.95 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 168.3, 160.4, 134.0, 124.8, 123.3, 120.3, 115.6, 50.1, 43.2, 29.4, 29.2, 26.4, 26.2, 26.2, 18.0. HRMS (ESI) calculated for C15H21ClNO2, 282.1261 (M + H)+; found, 282.1257.
5-Chloro-2-hydroxy-N-((tetrahydro-2H-pyran-4-yl)methyl)benzamide (18). Compound 18 (117 mg, 80%) was prepared as a light-yellow solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and 4-(aminomethyl)tetrahydropyran. HPLC purity 99.8% (tR = 16.12 min). 1H NMR (300 MHz, CDCl3) δ 12.23 (s, 1H), 7.45 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 9.0, 2.4 Hz, 1H), 7.06 (s, 1H), 6.91 (d, J = 9.0 Hz, 1H), 4.05–3.94 (m, 2H), 3.49–3.27 (m, 4H), 1.99–1.81 (m, 1H), 1.73–1.61 (m, 2H), 1.47–1.29 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.1, 159.8, 134.0, 125.5, 123.5, 120.0, 115.6, 67.6 (2C), 45.4, 35.2, 30.6 (2C). HRMS (ESI) calculated for C13H17ClNO3, 270.0897 (M + H)+; found, 270.0890.
tert-Butyl 4-((5-chloro-2-hydroxybenzamido)methyl)piperidine-1-carboxylate (19). Compound 19 (74 mg, 37%) was prepared as an off-white solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and 4-(aminomethyl)-1-N-Boc-piperidine. HPLC purity 99.8% (tR = 18.83 min). 1H NMR (300 MHz, CDCl3) δ 12.23 (s, 1H), 7.44 (d, J = 2.1 Hz, 1H), 7.30 (dd, J = 8.7, 2.1 Hz, 1H), 7.08–6.86 (m, 2H), 4.20–4.02 (m, 2H), 3.32 (s, 2H), 2.79–2.59 (m, 2H), 1.87–1.67 (m, 3H), 1.45 (s, 9H), 1.25–1.09 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.2, 160.1, 155.0, 134.1, 125.5, 123.5, 120.1, 115.6, 79.8, 45.2, 43.7 (2C), 36.4, 30.0 (2C), 28.6 (3C). HRMS (ESI) calculated for C18H26ClN2O4, 369.1581 (M + H)+; found, 369.1572.
5-Chloro-N-(2-cyclohexylethyl)-2-hydroxybenzamide (20). Compound 20 (69 mg, 46%) was prepared as an off-white solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and 2-cyclohexylethylamine. HPLC purity 99.9% (tR = 20.07 min). 1H NMR (300 MHz, CDCl3) δ 12.33 (s, 1H), 7.38–7.27 (m, 2H), 6.91 (d, J = 8.7 Hz, 1H), 6.37 (s, 1H), 3.51–3.39 (m, 2H), 1.80–1.60 (m, 5H), 1.56–1.44 (m, 2H), 1.39–1.14 (m, 4H), 1.02–0.84 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.0, 160.1, 134.0, 125.1, 123.4, 120.2, 115.5, 37.9, 36.9, 35.5, 33.2 (2C), 26.5, 26.2 (2C). HRMS (ESI) calculated for C15H21ClNO2, 282.1261 (M + H)+; found, 282.1253.
tert-Butyl 4-(2-(5-chloro-2-hydroxybenzamido)ethyl)piperazine-1-carboxylate (21). Compound 21 (88 mg, 43%) was prepared as an off-white solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and 4-N-(2-aminoethyl)-1-N-Boc-piperazine. HPLC purity 98.4% (tR = 15.08 min). 1H NMR (300 MHz, CDCl3) δ 7.35–7.25 (m, 2H), 7.15 (s, 1H), 6.89 (d, J = 8.7 Hz, 1H), 3.56–3.39 (m, 6H), 2.59 (t, J = 6.0 Hz, 2H), 2.49–2.38 (m, 4H), 1.44 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 168.9, 160.1, 154.8, 134.0, 125.4, 123.3, 120.1, 115.5, 80.0, 56.3, 52.8 (2C), 43.8 (2C), 36.1, 28.5 (3C). HRMS (ESI) calculated for C18H27ClN3O4, 384.1690 (M + H)+; found, 384.1680.
5-Chloro-2-hydroxy-N-(2-(4-methylpiperazin-1-yl)ethyl)benzamide (22). Compound 22 (62 mg, 37%) was prepared as a pale solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and 2-(4-methylpiperazin-1-yl)ethylamine. HPLC purity 95.0% (tR = 11.25 min). 1H NMR (300 MHz, CDCl3) δ 9.45 (br s, 1H, 7.34 (d, J = 2.4 Hz, 1H), 7.29 (dd, J = 9.0, 2.7 Hz, 1H), 7.20 (s, 1H), 6.88 (d, J = 9.0 Hz, 1H), 3.53–3.44 (m, 2H), 2.70–2.33 (m, 10H), 2.29 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 168.7, 160.0, 133.9, 125.6, 123.3, 120.0, 115.7, 56.0, 55.2 (2C), 52.8 (2C), 46.1, 36.0. HRMS (ESI) calculated for C14H21ClN3O2, 298.1322 (M + H)+; found, 298.1314.
5-Chloro-2-hydroxy-N-(2-morpholinoethyl)benzamide (23). Compound 23 (60 mg, 39%) was prepared as a grey solid according to general procedure A (r.t., 96 h), starting from methyl 5-chloro-2-hydroxybenzoate and 4-(2-aminoethyl)morpholine. HPLC purity 99.6% (tR = 11.66 min). 1H NMR (300 MHz, CDCl3) δ 7.35–7.28 (m, 2H), 7.03 (s, 1H), 6.94–6.88 (m, 1H), 3.74 (t, J = 4.5 Hz, 4H), 3.51 (t, J = 5.7 Hz, 2H), 2.61 (t, J = 6.0 Hz, 2H), 2.51 (t, J = 4.5 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 168.9, 160.2, 134.1, 125.3, 123.3, 120.2, 115.4, 67.1 (2C), 56.5, 53.4 (2C), 35.8. HRMS (ESI) calculated for C13H18ClN2O3, 285.1006 (M + H)+; found, 285.0999.
5-Chloro-N-hexyl-2-hydroxybenzamide (24). Compound 24 (107 mg, 78%) was prepared as a white solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and hexylamine. HPLC purity 98.0% (tR = 19.72 min). 1H NMR (300 MHz, CDCl3) δ 12.33 (s, 1H), 7.35 (d, J = 2.4 Hz, 1H), 7.30 (dd, J = 8.7, 2.4 Hz, 1H), 6.90 (d, J = 8.7 Hz, 1H), 6.52 (s, 1H), 3.46–3.36 (s, 2H), 1.67–1.53 (m, 2H), 1.42–1.20 (m, 6H), 0.92–0.82 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 169.0, 160.0, 134.0, 125.2, 123.4, 120.1, 115.5, 40.1, 31.5, 29.4, 26.7, 22.6, 14.1. HRMS (ESI) calculated for C13H19ClNO2, 256.1104 (M + H)+; found, 256.1098.
tert-Butyl (4-(5-chloro-2-hydroxybenzamido)butyl)carbamate (25). Compound 25 (56 mg, 30%) was prepared as a white solid according to general procedure A (r.t., 48 h), starting from methyl 5-chloro-2-hydroxybenzoate and N-Boc-1,4-butanediamine. HPLC purity 98.5% (tR = 17.75 min). 1H NMR (300 MHz, CDCl3) δ 12.53 (s, 1H), 7.63 (s, 1H), 7.53 (s, 1H), 7.29 (dd, J = 8.7, 2.4 Hz, 1H), 6.90 (d, J = 8.7 Hz, 1H), 4.77 (s, 1H), 3.48 (q, J = 6.0 Hz, 2H), 3.16 (q, J = 6.3 Hz, 2H), 1.72–1.53 (m, 4H), 1.45 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 169.3, 160.2, 156.9, 133.9, 125.9, 123.4, 119.9, 115.6, 79.9, 39.9, 39.8, 28.6, 28.6 (3C), 25.2. HRMS (ESI) calculated for C16H24ClN2O4, 343.1425 (M + H)+; found, 343.1417.
(5-Chloro-2-hydroxyphenyl)(piperidin-1-yl)methanone (26). To a solution of 5-chloro-2-methoxybenzoic acid (210 mg, 1.13 mmol), piperidine (80 mg, 0.94 mmol) and DMAP (28 mg, 0.23 mmol) in DCM (20 mL) was added EDCI (433 mg, 2.26 mmol) at 0 °C. The resulting mixture was stirred at r.t. for 12 h and concentrated. The residue was purified by column chromatography to afford the amide intermediate (5-chloro-2-methoxyphenyl)(piperidin-1-yl)methanone (230 mg, 96%) as colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.25–7.09 (m, 2H), 6.77 (d, J = 8.7 Hz, 1H), 3.74 (s, 3H), 3.69–3.52 (m, 2H), 3.19–3.03 (m, 2H), 1.69–1.31 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 165.9, 153.8, 129.7, 127.8, 127.5, 125.7, 112.2, 55.8, 47.9, 42.5, 26.3, 25.5, 24.5.
The amide intermediate (5-chloro-2-methoxyphenyl)(piperidin-1-yl)methanone (230 mg, 0.91 mmol) was dissolved in DCM (50 mL), and then BBr3 (4.53 mL, 4.53 mmol, 1 M in DCM) was added at 0 °C. The mixture was stirred at r.t. for 2 h. The mixture was diluted with DCM, washed with H2O and brine, dried (Na2SO4) and concentrated. The residue was purified by preparative TLC to afford compound 26 (203 mg, 93%) as a white solid. HPLC purity 95.1% (tR = 15.13 min). 1H NMR (300 MHz, CDCl3) δ 9.57 (s, 1H), 7.26 (dd, J = 8.7, 2.7 Hz, 1H), 7.19 (d, J = 2.7 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H), 3.67–3.60 (m, 4H), 1.77–1.61 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 169.4, 157.5, 132.3, 127.8, 123.4, 119.6, 118.9, 47.0 (2C), 26.2 (2C), 24.6. HRMS (ESI) calculated for C12H15ClNO2, 240.0791 (M + H)+; found, 240.0786.
1-(4-(5-Chloro-2-hydroxybenzoyl)piperazin-1-yl)ethanone (27). Compound 27 was prepared using a procedure similar to that used to prepare compound 26 starting from 5-chlorosalicylic acid and 1-acetylpiperazine. The title compound was obtained (139 mg, 91% in two steps) as a yellow solid. HPLC purity 97.2% (tR = 12.32 min). 1H NMR (300 MHz, CDCl3) δ 9.51 (s, 1H), 7.23–7.14 (m, 2H), 6.86 (d, J = 8.7 Hz, 1H), 3.72–3.48 (m, 8H), 2.11 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 169.6, 168.9, 155.0, 132.0, 127.9, 124.2, 120.8, 118.8, 46.1, 45.3, 44.8, 41.5, 21.4. HRMS (ESI) calculated for C13H16ClN2O3, 283.0849 (M + H)+; found, 283.0843.
(5-Chloro-2-hydroxyphenyl)(4-methylpiperazin-1-yl)methanone (28). Compound 28 (39 mg, 28%) was prepared as a pale yellow solid solid according to general procedure A (60 °C, 96 h), starting from methyl 5-chloro-2-hydroxybenzoate and 1-methylpiperazine. HPLC purity 98.3% (tR = 10.32 min). 1H NMR (300 MHz, CDCl3) δ 7.23 (dd, J = 8.7, 2.7 Hz, 1H), 7.17 (d, J = 2.7 Hz, 1H), 6.87 (d, J = 8.7 Hz, 1H), 3.69 (t, J = 5.1 Hz, 4H), 2.45 (t, J = 5.1 Hz, 4H), 2.32 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 169.2, 156.7, 132.2, 127.8 (2C), 123.7, 119.4, 55.0 (2C), 46.0, 45.5 (2C). HRMS (ESI) calculated for C12H16ClN2O2, 255.0900 (M + H)+; found, 255.0895.
4.2. Biology
4.2.1. Cell Lines and Virus Strain
Human A549 (ATCC® CCL-185™), HFF (ATCC® SCRC-1041™) and HEK-293 (ATCC® CRL-1573™) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The 293 β5 stable cell line overexpressing the human β5 integrin subunit and the HAdV-5 ts1 mutant were kindly provided by Dr. Glen Nemerow [45]. The cell lines were propagated in Dulbecco’s modified Eagle medium (DMEM, Life Technologies/Thermo Fisher, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Omega Scientific, Tarzana, CA, USA), 10 mM HEPES, 100 units/mL penicillin, 4 mM L-glutamine, 100 μg/mL streptomycin, and 0.1 mM nonessential amino acids (complete DMEM). Wild-type HAdV-5 and HCMV (AD169) were from the ATCC. The HAdV-GFP used in this study is a replication-defective HAdV5 that contains a CMV promoter-driven enhanced green fluorescent protein (eGFP) reporter gene cassette in place of the E1/E3 regions [46]. HAdVs were propagated in 293 β5 cells and separated from the cellular lysate by cesium chloride density gradient centrifugation. We calculated virus concentration, in mg/mL, with the Bio-Rad Protein Assay (Bio-Rad Laboratories, CA, USA) and converted it to virus particles/mL (vp/mL) using 4 × 1012 vp/mg.
4.2.2. Plaque Assay
Compounds were measured at concentrations of 10 μM and in a dose−response assay ranging from 10 to 0.3 μM using low MOI infections (0.06 vp/cell) in a plaque assay. In brief, 293 β5 cells were seeded in 6-well plates at a density of 4 × 105 cells per well in duplicate for each condition. Cells were infected with HAdV5-GFP (0.06 vp/cell) and rocked for 2 h at 37 °C, when cells reached 80%–90% confluency. When the incubation was finished, the inoculum was removed followed by washing the cells once with PBS. The cells were then carefully overlaid with 4 mL/well of equal parts of 1.6% (water/vol) Difco Agar Noble (Becton, Dickinson & Co., Sparks, MD, USA) and 2× EMEM (Minimum Essential Medium Eagle, BioWhittaker, MD, USA) supplemented with 2×penicillin/streptomycin, 2× L-glutamine, and 10% FBS. The compounds were also added to the mixture in concentrations ranging from 10 to 0.3 μM. After incubation for 7 days at 37 °C, we scanned plates with a Typhoon FLA 9000 imager (GE Healthcare Life Sciences, MA, USA) and quantified plaques with ImageJ [47].
4.2.3. Cytotoxicity Assay
The cytotoxicity of these derivatives was tested by commercial kit AlamarBlue® (Ref. DAL1025, Invitrogen, MA, USA). A549 cells at a density of 5 × 103 cells per well in 96-well plates were seeded. Decreasing concentrations of each derivative (200 μM, 150 μM, 100 μM, 80 μM, 60 μM, 40 μM, 30 μM, 20 μM, 10 μM, 5 μM, 2.5 μM, 0 μM) were diluted in 100 μL of Dulbecco’s Modified Eagle Medium (DMEM). Cells were then incubated at 37 °C for 48 h according to the kit protocol. The cytotoxic concentration 50 (CC50) value was obtained by using the statistical package GraphPad Prism. This assay was performed in duplicate.
4.2.4. Virus Yield Reduction
A549 cells (1.5 × 105 cells/well in a 24-well plate) were incubated for 24 h in 500 μL of complete DMEM and then infected with wild-type HAdV5 (100 vp/cell) when more than 90% of confluency were observed. Infected cells were incubated for 48 h at 37 °C in 500 μL of complete DMEM containing niclosamide (3) at 5 μM and derivative 16 at 10-fold IC50 concentration obtained in the plaque assay or the same volume of DMSO (positive control). After 48 h, cells were harvested and subjected to three rounds of freeze/thaw. Serial dilutions of clarified lysates were titrated on A549 cells (3 × 104 cells/well), and we calculated TCID50 values using an endpoint dilution method [48].
4.2.5. DNA Quantification by Real-Time PCR
A549 cells (1.5 × 105 cells/well in a 24-well plate) were incubated for 24 h in 500 μL of complete DMEM and then infected with wild-type HAdV5 (100 vp/cell) when more than 90% of confluency were observed. Infected cells were incubated 24 h at 37 °C in 500 μL of complete DMEM containing niclosamide (3) at 5 μM concentration and compound 16 at 10-fold its IC50 concentration in the plaque assay or the same volume of DMSO (positive control). All samples were carried out in duplicate. After incubation for 24 h at 37 °C, DNA was purified from the cell lysate with the E.Z.N.A.® Tissue DNA Kit (Omega Bio-tek, Norcross, GA, USA) following the manufacturer’s instructions. TaqMan primers and probes for a common region of the HAdV5 hexon were designed with the GenScript Real-Time PCR (TaqMan) Primer Design software (GenScript, Leiden, Netherlands). Oligonucleotides sequences were: AQ1: 5′-GCC ACG GTG GGG TTT CTA AAC TT-3′; AQ2: 5′-GCC CCA GTG GTC TTA CAT GCA CAT-3′; Probe: 6-FAM-5′-TGC ACC AGA CCC GGG CTC AGG TAC TCC GA-3′-TAMRA. Real-time PCR mixtures contained 9.5 μL of the purified DNA, AQ1 and AQ2 at a concentration of 200 nM each and Probe at a concentration of 50 nM in a total volume of 25 μL. The PCR cycling protocol was 95 °C for 3 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was applied as internal control. Oligonucleotides sequences for GAPDH and conditions were previously reported by Rivera et al. [49].
For quantification, gene fragments from hexon and GAPDH were cloned into the pGEM-T Easy vector (Promega, MA, USA), and known concentrations of template were used to produce a standard curve in parallel for each experiment. All assays were done in thermal cycler LightCycler® 96 System (Roche, Basel, Switzerland).
4.2.6. Nuclear-Associated HAdV Genomes
The nuclear delivery of HAdV genomes was evaluated by real-time PCR following nuclear isolation from infected cells. 1 × 106 A549 cells in 6-well plates were infected with wild-type HAdV5 at MOI 2000 vp/cell in the presence of 10-fold IC50 concentration obtained in the plaque assay for compound 16, niclosamide (3) at 5 μM or the same volume of DMSO for positive control. Forty-five minutes after infection, A549 cells were trypsinized, collected, and then washed twice with PBS. After that, we separated cytoplasmic and nuclear fractions using a hypotonic buffer solution and NP-40 detergent and resuspended the cell pellet in 500 μL of 1 × hypotonic buffer (20 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2). After incubation for 15 min at 4 °C, 25 μL of NP-40 was added and the samples were vortexed. The homogenates were centrifuged for 10 min at 835g and 4 °C. After the removal of the cytoplasmic fraction (supernatant), HAdV DNA was separated from the nuclear fraction (pellet) and from the cytoplasmic fraction using the E.Z.N.A.® Tissue DNA Kit (Omega Bio-tek, Norcross, GA, USA).
4.2.7. Time of Addition Assay
The anti-HAdV effects of compounds 3 and 16 at different points were evaluated in a time-curve assay using 293 β5 cells (3 × 105 cells/well in corning black wall, clear bottom 96-well plates) that were infected with HAdV5-GFP (2000 vp/cell) in the presence of 5 μM of compound 3 and 10-fold IC50 concentration obtained in the plaque assay for derivative 16. Parallel samples of HAdV-5 were incubated with or without the selected compounds on ice for 1 h. Virus was then added to 293 β5 cells and incubated at 37 °C. Compounds 3 and 16 were added at the indicated time points before or during this incubation. After a total of 2 h at 37 °C, cells were incubated for an additional 48 h at 37 °C and 5% CO2 before analyzing GFP expression using the Typhoon 9410 imager (GE Healthcare Life Sciences, MA, USA) as above.
4.2.8. HAdV-Mediated Endosome Disruption
A549 cells (~20,000 cells/well) were incubated in DMEM without cysteine or methionine supplemented with 10% dialyzed FBS [DMEM (−)] in black 96-well plates for 1 h before infection. Threefold serial dilutions (0.45 ng to 1000 ng) of HAdV5, or HAdV-5 ts1 were preincubated with cells in the presence of 5 µM niclosamide (3), 10-fold the IC50 concentration obtained in the plaque assay for compound 16, or the same volume of DMSO (negative control) for one hour. The medium was then removed and replaced with 50 μL DMEM (−) containing 0.1 mg/mL α-sarcin (Santa Cruz Biotechnology, Dallas, TX, USA) and the virus and drug mixtures. After 2 h at 37 °C, the Click-iT HPG Alexa Fluor 488 Protein Synthesis Assay Kits (Invitrogen, MA, USA) were used to analyze protein synthesis according to the manufacturer’s instructions. The incorporation of the amino acid analog of methionine L-homopropargylglycine (HPG) containing Alexa Fluor 488 azide was measured using a Typhoon 9410 imager (GE Healthcare Life Sciences, MA, USA) and calculated subtracting the background level of the control well containing L-homopropargylglycine (HPG) and α-sarcin but not virus (100% incorporation).
4.2.9. Thermostability Assay
HAdV-GFP (37.5 ng) was incubated with compound 16 (50 µM) or the same volume of DMSO for 1 h in complete DMEM. Parallel samples were incubated for 10 min at the indicated temperatures, cooled to r.t., and added to A549 cells on ice to synchronize the infection. After the incubation, cells were washed twice with PBS 1x to remove the nonattached virus and the excess of compound 16 and DMSO. Cells were then incubated for 24 h at 37 °C and 5% CO2 before being visualized for GFP expression in an Olympus inverted microscope Model IX71 (Hamburg, Germany) and analyzed by capturing representative images of each condition using the CellSens Dimension platform (Olympus, Hamburg, Germany).
4.2.10. Statistical Analyses
One-way ANOVA tests (Dunnet method) were performed using the GraphPad Prism 6. A P value under 0.05 was considered a statistical significance. We pointed out this statistical significance with asterisk in graphs, the numbers of which indicate the level of significance (* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001).
Supplementary Materials
Supplementary Materials can be found at https://www.mdpi.com/1422-0067/22/4/1617/s1, Copies of 1H and 13C NMR spectra.
Author Contributions
J.X., J.B.-C., M.C.-L. and H.C. performed research. J.X., J.B.-C., J.Z. and J.S.-C. designed research. J.X., J.B.-C., M.C.-L., H.C., J.Z. and J.S.-C. analyzed data. J.X., J.B.-C., M.C.-L., H.C., Y.X., E.A.W., J.P., J.Z. and J.S.-C. wrote or contributed to the writing and revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by the UTMB Technology Commercialization Program, the John D. Stobo, M. D. Distinguished Chair Endowment Fund (to JZ) and John Sealy Memorial Endowment Fund at UTMB, Plan Nacional de I + D + i 2013–2016 and Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Economía, Industria y Competitividad, Spanish Network for Research in Infectious Diseases (REIPI RD16/0016/0009), cofinanced by the European Development Regional Fund “A way to achieve Europe”, Operative program Intelligent Growth 20142020, the Instituto de Salud Carlos III, Proyectos de Investigación en Salud (PI15/00489) and Proyectos de Desarrollo Tecnológico en Salud (DTS17/00130), the Spanish Adenovirus Network (AdenoNet, BIO2015/68990-REDT), and the program “Nicolás Monardes” (C-0059-2018) Servicio Andaluz de Salud, Junta de Andalucía. E.A.W. is supported by the National Institutes of Health (NIH) National Research Service Award (NRSA) F31 DA04551. E.A.W. and J.Z. have no financial connections with the above European funding sources, and thus there is no conflict of interest to disclose.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| allo-HSCT | allogenic hematopoietic stem cell transplant |
| BCV | brincidofovir |
| CAP | community-acquired pneumonia |
| CC50 | half-maximal cytotoxic concentration |
| DCM | dichloromethane |
| DMAP | 4-(dimethylamino)pyridine |
| DMSO | dimethyl sulfoxide |
| EDCI | 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride |
| EtOAc | ethyl acetate |
| HAdVs | human adenoviruses |
| HCMV | human cytomegalovirus |
| HPG | L-homopropargylglycine |
| HPLC | high-performance liquid chromatography |
| HRMS | high-resolution mass spectrometry |
| IC50 | half-maximal inhibitory concentration |
| p.i. | postinfection |
| SAR | structure–activity relationship |
| SOT | solid-organ transplant |
| TLC | thin layer chromatography |
| TMS | tetramethylsilane |
| UV | ultraviolet |
References
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular evolution of human adenoviruses. Sci. Rep. 2013, 3, 1812. [Google Scholar] [CrossRef]
- HAdV Working Group Human Adenovirus Working Group. Available online: http://hadvwg.gmu.edu/ (accessed on 22 January 2020).
- Echavarria, M. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 2008, 21, 704–715. [Google Scholar] [CrossRef]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Raybould, J.E.; Sastry, S.; De la Cruz, O. Respiratory viruses in transplant recipients: More than just a cold. Clinical syndromes and infection prevention principles. Int. J. Infect. Dis. 2017, 62, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Sandkovsky, U.; Vargas, L.; Florescu, D.F. Adenovirus: Current epidemiology and emerging approaches to prevention and treatment. Curr. Infect. Dis. Rep. 2014, 16, 416. [Google Scholar] [CrossRef] [PubMed]
- Sedláček, P.; Petterson, T.; Robin, M.; Sivaprakasam, P.; Vainorius, E.; Brundage, T.; Chandak, A.; Mozaffari, E.; Nichols, G.; Voigt, S. Incidence of adenovirus infection in hematopoietic stem cell transplantation recipients: Findings from the advance study. Biol. Blood Marrow Transplant. 2019, 25, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, S.; Rodríguez, O.; Estrella, B.; Sabin, L.L.; Sempértegui, F.; Hamer, D.H. Etiology of severe pneumonia in Ecuadorian children. PLoS ONE 2017, 12, e0171687. [Google Scholar] [CrossRef]
- Kajon, A.E.; Ison, M.G. Severe infections with human adenovirus 7d in 2 adults in family, Illinois, USA, 2014. Emerg. Infect. Dis. 2016, 22, 730–733. [Google Scholar] [CrossRef]
- Tan, D.; Fu, Y.; Xu, J.; Wang, Z.; Cao, J.; Walline, J.; Zhu, H.; Yu, X. Severe adenovirus community-acquired pneumonia in immunocompetent adults: Chest radiographic and CT findings. J. Thorac. Dis. 2016, 8, 848–854. [Google Scholar] [CrossRef]
- Tan, D.; Zhu, H.; Fu, Y.; Tong, F.; Yao, D.; Walline, J.; Xu, J.; Yu, X. Severe community-acquired pneumonia caused by human adenovirus in immunocompetent adults: A multicenter case series. PLoS ONE 2016, 11, e0151199. [Google Scholar] [CrossRef]
- Yoon, B.W.; Song, Y.G.; Lee, S.H. Severe community-acquired adenovirus pneumonia treated with oral ribavirin: A case report. BMC Res. Notes 2017, 10, 47. [Google Scholar] [CrossRef]
- Yu, H.X.; Zhao, M.M.; Pu, Z.H.; Wang, Y.Q.; Liu, Y. Clinical data analysis of 19 cases of community-acquired adenovirus pneumonia in immunocompetent adults. Int. J. Clin. Exp. Med. 2015, 8, 19051–19057. [Google Scholar]
- Lenaerts, L.; De Clercq, E.; Naesens, L. Clinical features and treatment of adenovirus infections. Rev. Med. Virol. 2008, 18, 357–374. [Google Scholar] [CrossRef]
- Martínez-Aguado, P.; Serna-Gallego, A.; Marrugal-Lorenzo, J.A.; Gómez-Marín, I.; Sánchez-Céspedes, J. Antiadenovirus drug discovery: Potential targets and evaluation methodologies. Drug Discov. Today 2015, 20, 1235–1242. [Google Scholar] [CrossRef]
- Wold, W.S.M.; Tollefson, A.E.; Ying, B.; Spencer, J.F.; Toth, K. Drug development against human adenoviruses and its advancement by Syrian hamster models. FEMS Microbiol. Rev. 2019, 43, 380–388. [Google Scholar] [CrossRef]
- Andersson, E.K.; Strand, M.; Edlund, K.; Lindman, K.; Enquist, P.-A.; Spjut, S.; Allard, A.; Elofsson, M.; Mei, Y.-F.; Wadell, G. Small-molecule screening using a whole-cell viral replication reporter gene assay identifies 2-{[2-(benzoylamino)benzoyl]amino}-benzoic acid as a novel antiadenoviral compound. Antimicrob. Agents Chemother. 2010, 54, 3871–3877. [Google Scholar] [CrossRef]
- Oberg, C.T.; Strand, M.; Andersson, E.K.; Edlund, K.; Tran, N.P.; Mei, Y.F.; Wadell, G.; Elofsson, M. Synthesis, biological evaluation, and structure-activity relationships of 2-[2-(benzoylamino)benzoylamino]benzoic acid analogues as inhibitors of adenovirus replication. J. Med. Chem. 2012, 55, 3170–3181. [Google Scholar] [CrossRef]
- Sanchez-Cespedes, J.; Moyer, C.L.; Whitby, L.R.; Boger, D.L.; Nemerow, G.R. Inhibition of adenovirus replication by a trisubstituted piperazin-2-one derivative. Antivir. Res. 2014, 108, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, J.; Martinez-Aguado, P.; Vega-Holm, M.; Serna-Gallego, A.; Candela, J.I.; Marrugal-Lorenzo, J.A.; Pachon, J.; Iglesias-Guerra, F.; Vega-Perez, J.M. New 4-acyl-1-phenylaminocarbonyl-2-phenylpiperazine derivatives as potential inhibitors of adenovirus infection. Synthesis, biological evaluation, and structure-activity relationships. J. Med. Chem. 2016, 59, 5432–5448. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, S.; Marrugal-Lorenzo, J.A.; Vega-Holm, M.; Serna-Gallego, A.; Álvarez-Vidal, J.; Berastegui-Cabrera, J.; Pérez del Palacio, J.; Díaz, C.; Aiello, F.; Pachón, J.; et al. Optimization of piperazine-derived ureas privileged structures for effective antiadenovirus agents. Eur. J. Med. Chem. 2020, 185, 111840. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.M.; Sortino, K.; Sethna, P.; Bae, A.; Lanier, R.; Bambara, R.A.; Dewhurst, S. Cidofovir diphosphate inhibits adenovirus 5 DNA polymerase via both nonobligate chain termination and direct inhibition, and polymerase mutations confer cidofovir resistance on intact virus. Antimicrob. Agents Chemother. 2019, 63, e01925-18. [Google Scholar] [CrossRef]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antivir. Res. 2009, 82, 84–98. [Google Scholar] [CrossRef]
- Tippin, T.K.; Morrison, M.E.; Brundage, T.M.; Mommeja-Marin, H. Brincidofovir is not a substrate for the human organic anion transporter 1: A mechanistic explanation for the lack of nephrotoxicity observed in clinical studies. Ther. Drug Monit. 2016, 38, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Spencer, J.F.; Dhar, D.; Sagartz, J.E.; Buller, R.M.; Painter, G.R.; Wold, W.S. Hexadecyloxypropyl-cidofovir, CMX001, prevents adenovirus-induced mortality in a permissive, immunosuppressed animal model. Proc. Natl. Acad. Sci. USA 2008, 105, 7293–7297. [Google Scholar] [CrossRef] [PubMed]
- Grimley, M.S.; Marón, G.M.; Prasad, V.K.; Jacobsohn, D.A.; Young, J.-A.H.; Chittick, G.; Brundage, T.M.; Momméja-Marin, H. Preliminary results from the advise study evaluating brincidofovir (CMX001, BCV) for the treatment of disseminated and high-risk adenovirus (AdV) infection. Biol. Blood Marrow Transplant. 2015, 21, 108–109. [Google Scholar] [CrossRef]
- Prasad, V.K.; Papanicolaou, G.A.; Marón, G.M.; Vainorius, E.; Brundage, T.M.; Chittick, G.; Nichols, G.; Grimley, M.S. 46-Treatment of adenovirus (AdV) infection in allogeneic hematopoietic cell transplant (allo HCT) patients (pts) with brincidofovir: Final 36 week results from the advise trial. Biol. Blood Marrow Transplant. 2017, 23, 57–58. [Google Scholar] [CrossRef]
- Chittick, G.; Morrison, M.; Brundage, T.; Nichols, W.G. Short-term clinical safety profile of brincidofovir: A favorable benefit–risk proposition in the treatment of smallpox. Antivir. Res. 2017, 143, 269–277. [Google Scholar] [CrossRef]
- Grimley, M.S.; Chemaly, R.F.; Englund, J.A.; Kurtzberg, J.; Chittick, G.; Brundage, T.M.; Bae, A.; Morrison, M.E.; Prasad, V.K. Brincidofovir for asymptomatic adenovirus viremia in pediatric and adult allogeneic hematopoietic cell transplant recipients: A randomized placebo-controlled phase II trial. Biol. Blood Marrow Transplant. 2017, 23, 512–521. [Google Scholar] [CrossRef]
- Fan, X.; Xu, J.; Files, M.; Cirillo, J.D.; Endsley, J.J.; Zhou, J.; Endsley, M.A. Dual activity of niclosamide to suppress replication of integrated HIV-1 and Mycobacterium tuberculosis (Beijing). Tuberculosis 2019, 116, 28–33. [Google Scholar] [CrossRef]
- Xu, J.; Xie, X.; Ye, N.; Zou, J.; Chen, H.; White, M.A.; Shi, P.Y.; Zhou, J. Design, synthesis, and biological evaluation of substituted 4,6-dihydrospiro[[1,2,3]triazolo[4,5-b]pyridine-7,3’-indoline]-2’,5(3H)-dione analogues as potent NS4B inhibitors for the treatment of dengue virus infection. J. Med. Chem. 2019, 62, 7941–7960. [Google Scholar] [CrossRef]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Chen, H.; Wold, E.A.; Shi, P.Y.; Zhou, J. Therapeutic potential of spirooxindoles as antiviral agents. ACS Infect. Dis. 2016, 2, 382–392. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.-Y.; Li, H.; Zhou, J. Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, J.; Lang, Y.; Fan, X.; Kuo, L.; D’Brant, L.; Hu, S.; Samrat, S.K.; Trudeau, N.; Tharappel, A.M.; et al. JMX0207, a niclosamide derivative with improved pharmacokinetics, suppresses ZIKA virus infection both in vitro and in vivo. ACS Infect. Dis. 2020, 6, 2616–2628. [Google Scholar] [CrossRef]
- Xu, J.; Xie, X.; Chen, H.; Zou, J.; Xue, Y.; Ye, N.; Shi, P.-Y.; Zhou, J. Design, synthesis and biological evaluation of spiropyrazolopyridone derivatives as potent dengue virus inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127162. [Google Scholar] [CrossRef]
- Marrugal-Lorenzo, J.A.; Serna-Gallego, A.; Berastegui-Cabrera, J.; Pachón, J.; Sánchez-Céspedes, J. Repositioning salicylanilide anthelmintic drugs to treat adenovirus infections. Sci. Rep. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Berastegui-Cabrera, J.; Chen, H.; Pachón, J.; Zhou, J.; Sánchez-Céspedes, J. Structure–activity relationship studies on diversified salicylamide derivatives as potent inhibitors of human adenovirus infection. J. Med. Chem. 2020, 63, 3142–3160. [Google Scholar] [CrossRef]
- Xu, J.; Berastegui-Cabrera, J.; Ye, N.; Carretero-Ledesma, M.; Pachón-Díaz, J.; Chen, H.; Pachón-Ibáñez, M.E.; Sánchez-Céspedes, J.; Zhou, J. Discovery of novel substituted N-(4-Amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues as potent human adenovirus inhibitors. J. Med. Chem. 2020, 63, 12830–12852. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef]
- Rancourt, C.; Keyvani-Amineh, H.; Sircar, S.; Labrecque, P.; Weber, J.M. Proline 137 is critical for adenovirus protease encapsidation and activation but not enzyme activity. Virology 1995, 209, 167–173. [Google Scholar] [CrossRef]
- Smith, J.G.; Nemerow, G.R. Mechanism of adenovirus neutralization by Human α-defensins. Cell Host Microbe 2008, 3, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Luisoni, S.; Suomalainen, M.; Boucke, K.; Tanner, L.B.; Wenk, M.R.; Guan, X.L.; Grzybek, M.; Coskun, Ü.; Greber, U.F. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe 2015, 18, 75–85. [Google Scholar] [CrossRef]
- Matthews, H.; Deakin, J.; Rajab, M.; Idris-Usman, M.; Nirmalan, N.J. Investigating antimalarial drug interactions of emetine dihydrochloride hydrate using CalcuSyn-based interactivity calculations. PLoS ONE 2017, 12, e0173303. [Google Scholar] [CrossRef]
- Nguyen, E.K.; Nemerow, G.R.; Smith, J.G. Direct evidence from single-cell analysis that human α-defensins block adenovirus uncoating to neutralize infection. J. Virol. 2010, 84, 4041–4049. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno, R.R.; Pache, L.; Nemerow, G.R. Enhancement of gene transfer to human myeloid cells by adenovirus-fiber complexes. Mol. Ther. 2007, 15, 571–578. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Meth. 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Rivera, A.A.; Wang, M.; Suzuki, K.; Uil, T.G.; Krasnykh, V.; Curiel, D.T.; Nettelbeck, D.M. Mode of transgene expression after fusion to early or late viral genes of a conditionally replicating adenovirus via an optimized internal ribosome entry site in vitro and in vivo. Virology 2004, 320, 121–134. [Google Scholar] [CrossRef]





























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).