
The Roles of Post-Translational Modifications on mTOR Signaling

Abstract
:1. Introduction
2. Roles of PTMs on Shared Components of the mTOR Signaling
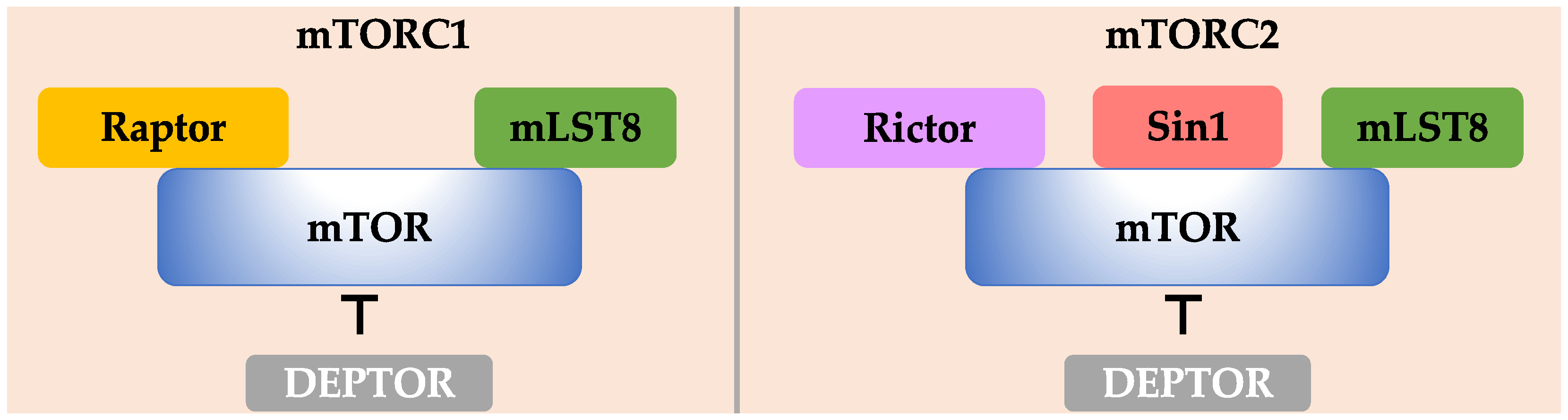
2.1. mTOR
2.2. mLST8
2.3. DEPTOR
3. Regulation of Components in mTORC1 Pathway by PTMs
3.1. Raptor
3.2. PRAS40
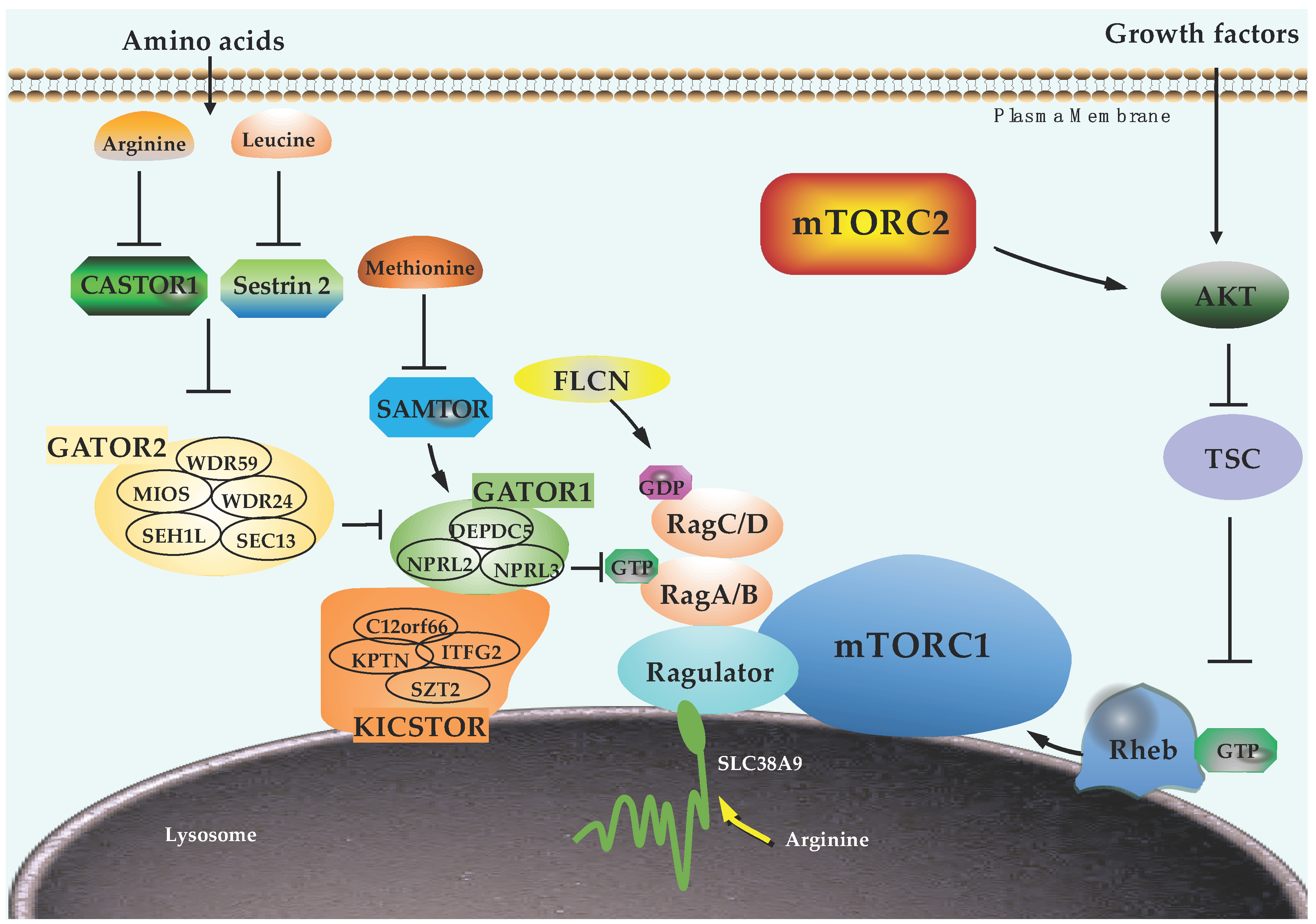
3.3. TSC Complex
3.4. Rheb GTPase
3.5. Rag GTPases
3.6. Ragulator Complex
3.7. GATOR1, GATOR2, and KICSTOR Complexes
3.8. FLCN
3.9. Amino Acid Sensors
3.10. mTORC1 Downstream Targets
4. Regulation of Components in mTORC2 Pathway by PTMs
4.1. Rictor
4.2. Sin1
4.3. PI3K and PTEN
4.4. mTORC2-Specific GTPases
4.5. mTORC2 Downstream Effectors
5. Targeting mTOR Signaling for Treating Human Diseases
5.1. mTOR Specific Inhibitors
5.2. Dual PI3K/mTOR Inhibitors
5.3. Akt Inhibitors
5.4. Targeting mTOR Signaling Regulators
5.4.1. Kinases
5.4.2. E3 Ubiquitin Ligases and Deubiquitinases (DUBs)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4E-BPs | eukaryotic initiation factor 4E-binding proteins |
| AMBRA1 | autophagy and Beclin 1 regulator 1 |
| AMPK | AMP-activated protein kinase |
| ATF4 | activating transcription factor 4 |
| ATG13 | Autophagy-related protein 13 |
| ATXN3 | Ataxin-3 |
| CAD | carbamoyl-phosphate synthetase 2 |
| CASTOR1 | cellular arginine sensor for mTORC1 |
| CDC25 | cell division cycle 25 |
| CHIP | carboxy-terminus of Hsc70 interacting protein |
| CRBN | cereblon |
| CUL4 | cullin 4 |
| DCAF15 | DDB1- and CUL4-associated factor 15 |
| DDB1 | DNA damage-binding protein 1 |
| DEPDC5 | DEP domain-containing 5 |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| E6AP | E6-associated protein |
| EP300 | E1A Binding Protein P300 |
| ERK | extracellular signal-regulated kinase |
| FASN | fatty acid synthase |
| FBXW7 | F-box and WD repeat domain containing 7 |
| FDA | Food and Drug Administration |
| FLCN | folliculin |
| FNIP | folliculin interacting protein |
| GATOR | GTPase activating protein (GAP) activity toward Rags |
| Grb10 | growth factor receptor-bound protein 10 |
| GSK3 | glycogen synthase kinase 3 |
| HBXIP | hepatitis B virus X-interacting protein |
| IAPs | inhibitors of apoptosis proteins |
| IKKβ | IkappaB kinase beta |
| IRFs | interferon regulatory factors |
| JNKs | c-Jun N-terminal protein kinases |
| KICSTOR | KPTN, ITFG2, C12orf66 and SZT2-containing regulator of mTORC1 |
| KLHL22 | Kelch-like protein 22 |
| LATS | Large tumor suppressor kinase |
| MAPK | mitogen-activated protein kinase |
| MCL1 | myeloid cell leukemia 1 |
| MEFs | mouse embryonic fibroblasts |
| MiT-TFE | microphthalmia/transcription factor E |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase 2 |
| NEDD4L | neural precursor cell expressed developmentally downregulated gene 4-like |
| NPRL2 | Nitrogen permease regulator 2-like protein |
| NPRL3 | Nitrogen permease regulator 3-like protein |
| NSCLC | Non-small-cell lung carcinoma |
| O-GlcNAc | O-Linked β-N-acetylglucosamine |
| OGT | O-linked N-acetylglucosamine transferase |
| OTUD7B | ovarian tumor domain-containing protein 7B |
| Pam | protein associated with Myc |
| PCAF | P300/CBP-associated factor |
| PDK1 | phosphoinositide dependent protein Kkinase 1 |
| PI3K | Phosphoinositide 3-kinase |
| PKA | protein kinase A |
| PKC | Protein kinase C |
| PKM2 | pyruvate kinase M2 |
| PPARγ | peroxisome proliferator-activated receptor-gamma |
| PRAK | p38 regulated/activated kinase |
| PRAS40 | proline-rich Akt substrate of 40 kDa |
| PTEN | phosphatase and tensin homolog |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| Rap1 | Ras-related protein 1 |
| Raptor | regulatory-associated protein of mTOR |
| Rheb | Ras homolog enriched in brain |
| PKB | Protein kinase B |
| Rictor | rapamycin insensitive companion of mTOR |
| RNF114 | ring finger protein 114 |
| RNF152 | ring Finger Protein 152 |
| RNF186 | ring finger protein 186 |
| RSKs | 90 kDa ribosomal S6 kinases |
| S6K | ribosomal protein S6 kinase |
| SAMTOR | S-adenosylmethionine sensor upstream of mTORC1 |
| SGK1 | serum and glucocorticoid-regulated kinase 1 |
| Sin1 | SAPK interacting protein 1 |
| Skp2 | S-phase kinase associated protein 2 |
| SREBP1/2 | transcriptional factors sterol regulatory element binding protein 1/2 |
| TFEB | transcription factor EB |
| TRAF2 | tumor necrosis factor receptor-associated factor 2 |
| TRAF6 | tumor necrosis factor receptor-associated factor 6 |
| TRIM31 | tripartite motif-containing protein 31 |
| TSC | Tuberous sclerosis complex |
| UCH-L1 | ubiquitin carboxy-terminal hydrolase L1 |
| ULK1 | unc-51 like autophagy activating kinase 1 |
| UVRAG | UV radiation resistance-associated gene protein |
| VHL | Von Hippel-Lindau |
| WDR24 | WD repeat domain 24 |
| WDR59 | WD repeat domain 59 |
References
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Proud, C.G. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006, 21, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 2014, 159, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Senoo, H.; Kamimura, Y.; Kimura, R.; Nakajima, A.; Sawai, S.; Sesaki, H.; Iijima, M. Phosphorylated Rho-GDP directly activates mTORC2 kinase towards AKT through dimerization with Ras-GTP to regulate cell migration. Nat. Cell Biol. 2019, 21, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef] [Green Version]
- Holz, M.K.; Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, B.T.; Ouwens, M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 1999, 344 Pt 2, 427–431. [Google Scholar] [CrossRef]
- Sekulic, A.; Hudson, C.C.; Homme, J.L.; Yin, P.; Otterness, D.M.; Karnitz, L.M.; Abraham, R.T. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000, 60, 3504–3513. [Google Scholar] [PubMed]
- Cheng, S.W.; Fryer, L.G.; Carling, D.; Shepherd, P.R. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J. Biol. Chem. 2004, 279, 15719–15722. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Jaquez, H.A.; Keller, J.A.; Foster, K.G.; Ekim, B.; Soliman, G.A.; Feener, E.P.; Ballif, B.A.; Fingar, D.C. Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol. Cell. Biol. 2009, 29, 4308–4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, R.T.; Beal, P.A.; Comb, M.J.; Schreiber, S.L. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J. Biol. Chem. 2000, 275, 7416–7423. [Google Scholar] [CrossRef] [Green Version]
- Copp, J.; Manning, G.; Hunter, T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): Phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009, 69, 1821–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, G.A.; Acosta-Jaquez, H.A.; Dunlop, E.A.; Ekim, B.; Maj, N.E.; Tee, A.R.; Fingar, D.C. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J. Biol. Chem. 2010, 285, 7866–7879. [Google Scholar] [CrossRef] [Green Version]
- Ekim, B.; Magnuson, B.; Acosta-Jaquez, H.A.; Keller, J.A.; Feener, E.P.; Fingar, D.C. mTOR kinase domain phosphorylation promotes mTORC1 signaling, cell growth, and cell cycle progression. Mol. Cell. Biol. 2011, 31, 2787–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Lee, H. Phosphorylated mTOR Expression Profiles in Human Normal and Carcinoma Tissues. Dis. Markers 2017, 2017, 1397063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Tian, C.; Sun, J.; Zhang, J.; Ren, K.; Fan, X.Y.; Wang, K.; Wang, H.; Yan, Y.E.; Chen, C.; et al. FBXW7-Induced MTOR Degradation Forces Autophagy to Counteract Persistent Prion Infection. Mol. Neurobiol. 2016, 53, 706–719. [Google Scholar] [CrossRef]
- Park, D.; Lee, M.N.; Jeong, H.; Koh, A.; Yang, Y.R.; Suh, P.G.; Ryu, S.H. Parkin ubiquitinates mTOR to regulate mTORC1 activity under mitochondrial stress. Cell Signal. 2014, 26, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.; Kim, L.C.; Song, W.; Edwards, D.N.; Cook, R.S.; Chen, J. Disruption of the Scaffolding Function of mLST8 Selectively Inhibits mTORC2 Assembly and Function and Suppresses mTORC2-Dependent Tumor Growth In Vivo. Cancer Res. 2019, 79, 3178–3184. [Google Scholar] [CrossRef]
- Wang, B.; Jie, Z.; Joo, D.; Ordureau, A.; Liu, P.; Gan, W.; Guo, J.; Zhang, J.; North, B.J.; Dai, X.; et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 2017, 545, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhong, J.; Inuzuka, H.; Gao, D.; Shaik, S.; Sarkar, F.H.; Wei, W. An evolving role for DEPTOR in tumor development and progression. Neoplasia 2012, 14, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Skaar, J.R.; Kuchay, S.; Toschi, A.; Kanarek, N.; Ben-Neriah, Y.; Pagano, M. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol. Cell 2011, 44, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Inuzuka, H.; Tan, M.K.; Fukushima, H.; Locasale, J.W.; Liu, P.; Wan, L.; Zhai, B.; Chin, Y.R.; Shaik, S.; et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 2011, 44, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Xiong, X.; Sun, Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 2011, 44, 304–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Teran, B.; Lopez, J.A.; Rodriguez, E.; Leiva, L.; Martinez-Martinez, S.; Bernal, J.A.; Jimenez-Borreguero, L.J.; Redondo, J.M.; Vazquez, J.; Sabio, G. p38gamma and delta promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat. Commun. 2016, 7, 10477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Ruegg, M.A.; et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 2011, 123, 1073–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.L.; Tang, Y.; Li, H.; Guertin, D.A. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab. 2016, 5, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Romanino, K.; Cloetta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Umemura, A.; Park, E.J.; Taniguchi, K.; Lee, J.H.; Shalapour, S.; Valasek, M.A.; Aghajan, M.; Nakagawa, H.; Seki, E.; Hall, M.N.; et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 2014, 20, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J. Biol. Chem. 2010, 285, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Fujishita, T.; Aoki, M.; Taketo, M.M. JNK signaling promotes intestinal tumorigenesis through activation of mTOR complex 1 in Apc(Delta716) mice. Gastroenterology 2011, 140, 1556–1563.e6. [Google Scholar] [CrossRef] [PubMed]
- Kwak, D.; Choi, S.; Jeong, H.; Jang, J.H.; Lee, Y.; Jeon, H.; Lee, M.N.; Noh, J.; Cho, K.; Yoo, J.S.; et al. Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal Kinase (JNK)-mediated Raptor protein phosphorylation. J. Biol. Chem. 2012, 287, 18398–18407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Valle, F.; Badura, M.L.; Braunstein, S.; Narasimhan, M.; Schneider, R.J. Mitotic raptor promotes mTORC1 activity, G(2)/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol. Cell. Biol. 2010, 30, 3151–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Asara, J.M.; Shaw, R.J. Raptor is phosphorylated by cdc2 during mitosis. PLoS ONE 2010, 5, e9197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriere, A.; Cargnello, M.; Julien, L.A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P.P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, W.; Dai, X.; Dai, X.; Xie, J.; Yin, S.; Zhu, J.; Wang, C.; Liu, Y.; Guo, J.; Wang, M.; et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat. Cell Biol. 2020, 22, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Wu, M.; Zhang, H.; Sun, H. mTORC1 signaling requires proteasomal function and the involvement of CUL4-DDB1 ubiquitin E3 ligase. Cell Cycle 2008, 7, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Feldman, A.L.; Das, C.; Ziesmer, S.C.; Ansell, S.M.; Galardy, P.J. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol. Cell. Biol. 2013, 33, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jeno, P.; Arrieumerlou, C.; Hall, M.N. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE 2007, 2, e1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Lv, D.; Guo, L.; Zhang, T.; Huang, L. PRAS40 signaling in tumor. Oncotarget 2017, 8, 69076–69085. [Google Scholar] [CrossRef] [Green Version]
- Madhunapantula, S.V.; Sharma, A.; Robertson, G.P. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 2007, 67, 3626–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shipitsin, M.; Small, C.; Giladi, E.; Siddiqui, S.; Choudhury, S.; Hussain, S.; Huang, Y.E.; Chang, H.; Rimm, D.L.; Berman, D.M.; et al. Automated quantitative multiplex immunofluorescence in situ imaging identifies phospho-S6 and phospho-PRAS40 as predictive protein biomarkers for prostate cancer lethality. Proteome Sci. 2014, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.Z.; Deng, A.M.; Li, L.H.; Liu, G.Y.; Wu, G.Y. Prognostic role of phospho-PRAS40 (Thr246) expression in gastric cancer. Arch. Med. Sci. 2014, 10, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, E.E.; Elder, D.J.; Thomas, E.C.; Phillips, L.; Morgan, C.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavare, J.M. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Jazet, I.M.; Schaart, G.; Gastaldelli, A.; Ferrannini, E.; Hesselink, M.K.; Schrauwen, P.; Romijn, J.A.; Maassen, J.A.; Pijl, H.; Ouwens, D.M.; et al. Loss of 50% of excess weight using a very low energy diet improves insulin-stimulated glucose disposal and skeletal muscle insulin signalling in obese insulin-treated type 2 diabetic patients. Diabetologia 2008, 51, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Tang, S.; Zhang, X.; Chen, L. Targeting PRAS40: A novel therapeutic strategy for human diseases. J. Drug Target. 2021, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlova, K.A.; Crino, P.B. The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 2010, 1184, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballif, B.A.; Roux, P.P.; Gerber, S.A.; MacKeigan, J.P.; Blenis, J.; Gygi, S.P. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Ma, X.; Zhao, W.; Huai, W.; Li, T.; Qiu, Y.; Zhang, Y.; Han, L. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene 2018, 37, 478–488. [Google Scholar] [CrossRef]
- Han, S.; Witt, R.M.; Santos, T.M.; Polizzano, C.; Sabatini, B.L.; Ramesh, V. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal. 2008, 20, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zacharek, S.; He, Y.J.; Lee, H.; Shumway, S.; Duronio, R.J.; Xiong, Y. WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev. 2008, 22, 866–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, G.J.; Kinch, M.S.; Rogers-Graham, K.; Sebti, S.M.; Hamilton, A.D.; Der, C.J. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J. Biol. Chem. 1997, 272, 10608–10615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Nakagawa, M.; Young, S.G.; Yamanaka, S. Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J. Biol. Chem. 2005, 280, 32768–32774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Wang, Y.H.; Wu, X.N.; Wu, S.Q.; Lu, B.J.; Dong, M.Q.; Zhang, H.; Sun, P.; Lin, S.C.; Guan, K.L.; et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Hong, S.; Ikeda, T.; Mori, H.; MacDougald, O.A.; Nada, S.; Okada, M.; Inoki, K. Amino Acids Enhance Polyubiquitination of Rheb and Its Binding to mTORC1 by Blocking Lysosomal ATXN3 Deubiquitinase Activity. Mol. Cell 2020, 80, 437–451.e6. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Zhu, H.; Silva, R.L.; Mills, J.R.; Teruya-Feldstein, J.; Lowe, S.W.; Tam, W.; Pelletier, J.; Wendel, H.G. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008, 22, 2178–2188. [Google Scholar] [CrossRef] [Green Version]
- Potheraveedu, V.N.; Schopel, M.; Stoll, R.; Heumann, R. Rheb in neuronal degeneration, regeneration, and connectivity. Biol. Chem. 2017, 398, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat. Genet 2016, 48, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, P.A.; Zimmermann, M.T.; Kim, M.; Evans, J.M.; Xu, X.; Olson, T.M. De novo RRAGC mutation activates mTORC1 signaling in syndromic fetal dilated cardiomyopathy. Hum. Genet. 2016, 135, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Jiang, C.; Chen, L.; Jin, J.; Wei, J.; Zhao, L.; Chen, M.; Pan, W.; Xu, Y.; Chu, H.; et al. The ubiquitination of rag A GTPase by RNF152 negatively regulates mTORC1 activation. Mol. Cell 2015, 58, 804–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Lee, S.W.; Zhang, X.; Cai, Z.; Gao, Y.; Chou, P.C.; Rezaeian, A.H.; Han, F.; Wang, C.Y.; Yao, J.C.; et al. Skp2-Mediated RagA Ubiquitination Elicits a Negative Feedback to Prevent Amino-Acid-Dependent mTORC1 Hyperactivation by Recruiting GATOR1. Mol. Cell 2015, 58, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Humphrey, S.J.; Murashige, D.S.; Francis, D.; Wang, Q.P.; Cooke, K.C.; Neely, G.G.; James, D.E. RagC phosphorylation autoregulates mTOR complex 1. EMBO J. 2019, 38, e99548. [Google Scholar] [CrossRef]
- Nada, S.; Hondo, A.; Kasai, A.; Koike, M.; Saito, K.; Uchiyama, Y.; Okada, M. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009, 28, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Liu, Y.; Jia, Y.; Hao, X.; Lin, W.J.; Tran, J.; Lynch, G.; Baudry, M.; Bi, X. UBE3A-mediated p18/LAMTOR1 ubiquitination and degradation regulate mTORC1 activity and synaptic plasticity. Elife 2018, 7, e37993. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.A.; Zori, R.T.; Stone, J.W.; Gray, B.A.; Cantu, E.S.; Ostrer, H. Maternal origin of 15q11-13 deletions in Angelman syndrome suggests a role for genomic imprinting. Am. J. Med. Genet. 1990, 35, 350–353. [Google Scholar] [CrossRef]
- Cook, E.H., Jr.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997, 60, 928–934. [Google Scholar] [PubMed]
- Rasheed, N.; Lima, T.B.; Mercaldi, G.F.; Nascimento, A.F.Z.; Silva, A.L.S.; Righetto, G.L.; Bar-Peled, L.; Shen, K.; Sabatini, D.M.; Gozzo, F.C.; et al. C7orf59/LAMTOR4 phosphorylation and structural flexibility modulate Ragulator assembly. FEBS Open Bio 2019, 9, 1589–1602. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ou, Y.; Yang, Y.; Li, W.; Xu, Y.; Xie, Y.; Liu, Y. KLHL22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature 2018, 557, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Padi, S.K.R.; Singh, N.; Bearss, J.J.; Olive, V.; Song, J.H.; Cardo-Vila, M.; Kraft, A.S.; Okumura, K. Phosphorylation of DEPDC5, a component of the GATOR1 complex, releases inhibition of mTORC1 and promotes tumor growth. Proc. Natl. Acad. Sci. USA 2019, 116, 20505–20510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldassari, S.; Picard, F.; Verbeek, N.E.; van Kempen, M.; Brilstra, E.H.; Lesca, G.; Conti, V.; Guerrini, R.; Bisulli, F.; Licchetta, L.; et al. The landscape of epilepsy-related GATOR1 variants. Genet. Med. 2019, 21, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basel-Vanagaite, L.; Hershkovitz, T.; Heyman, E.; Raspall-Chaure, M.; Kakar, N.; Smirin-Yosef, P.; Vila-Pueyo, M.; Kornreich, L.; Thiele, H.; Bode, H.; et al. Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am. J. Hum. Genet. 2013, 93, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; Duray, P.; Merino, M.; Choyke, P.; Pavlovich, C.P.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2002, 2, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Hasumi, Y.; Baba, M.; Hasumi, H.; Huang, Y.; Lang, M.; Reindorf, R.; Oh, H.B.; Sciarretta, S.; Nagashima, K.; Haines, D.C.; et al. Folliculin (Flcn) inactivation leads to murine cardiac hypertrophy through mTORC1 deregulation. Hum. Mol. Genet. 2014, 23, 5706–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villen, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Seifan, S.; Claessens, T.; Behrends, C.; Kamps, M.A.; Rozycka, E.; Kemp, A.J.; Nookala, R.K.; Blenis, J.; Coull, B.J.; et al. FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy 2014, 10, 1749–1760. [Google Scholar] [CrossRef]
- Piao, X.; Kobayashi, T.; Wang, L.; Shiono, M.; Takagi, Y.; Sun, G.; Abe, M.; Hagiwara, Y.; Zhang, D.; Okimoto, K.; et al. Regulation of folliculin (the BHD gene product) phosphorylation by Tsc2-mTOR pathway. Biochem. Biophys. Res. Commun. 2009, 389, 16–21. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, J.M.; Sylvestersen, K.B.; Bekker-Jensen, S.; Szklarczyk, D.; Poulsen, J.W.; Horn, H.; Jensen, L.J.; Mailand, N.; Nielsen, M.L. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell. Proteom. 2011, 10, M110.003590. [Google Scholar] [CrossRef] [Green Version]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jeon, B.T.; Kim, I.M.; Bennett, S.J.; Lorch, C.M.; Viana, M.P.; Myers, J.F.; Trupp, C.J.; Whipps, Z.T.; Kundu, M.; et al. Sestrin2 Phosphorylation by ULK1 Induces Autophagic Degradation of Mitochondria Damaged by Copper-Induced Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 6130. [Google Scholar] [CrossRef] [PubMed]
- Kimball, S.R.; Gordon, B.S.; Moyer, J.E.; Dennis, M.D.; Jefferson, L.S. Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation. Cell Signal. 2016, 28, 896–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lear, T.B.; Lockwood, K.C.; Ouyang, Y.; Evankovich, J.W.; Larsen, M.B.; Lin, B.; Liu, Y.; Chen, B.B. The RING-type E3 ligase RNF186 ubiquitinates Sestrin-2 and thereby controls nutrient sensing. J. Biol. Chem. 2019, 294, 16527–16534. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, M.; Goyette, P.; Boucher, G.; Lo, K.S.; Rivas, M.A.; Stevens, C.; Alikashani, A.; Ladouceur, M.; Ellinghaus, D.; Torkvist, L.; et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013, 9, e1003723. [Google Scholar] [CrossRef] [PubMed]
- Pasha, M.; Eid, A.H.; Eid, A.A.; Gorin, Y.; Munusamy, S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxid. Med. Cell Longev. 2017, 2017, 3296294. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, N.; Thomas, G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997, 410, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, N.; Dennis, P.B.; Andjelkovic, M.; Dufner, A.; Kozma, S.C.; Hemmings, B.A.; Thomas, G. Phosphorylation and activation of p70s6k by PDK1. Science 1998, 279, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Weng, Q.P.; Kozlowski, M.; Belham, C.; Zhang, A.; Comb, M.J.; Avruch, J. Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J. Biol. Chem. 1998, 273, 16621–16629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Zhao, B.; Lombard, D.B.; Fingar, D.C.; Inoki, K. Cross-talk between sirtuin and mammalian target of rapamycin complex 1 (mTORC1) signaling in the regulation of S6 kinase 1 (S6K1) phosphorylation. J. Biol. Chem. 2014, 289, 13132–13141. [Google Scholar] [CrossRef] [Green Version]
- Panasyuk, G.; Nemazanyy, I.; Filonenko, V.; Gout, I. Ribosomal protein S6 kinase 1 interacts with and is ubiquitinated by ubiquitin ligase ROC1. Biochem. Biophys. Res. Commun. 2008, 369, 339–343. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Pyo, K.E.; Kim, C.R.; Lee, M.; Kim, J.S.; Kim, K.I.; Baek, S.H. ULK1 O-GlcNAcylation Is Crucial for Activating VPS34 via ATG14L during Autophagy Initiation. Cell Rep. 2018, 25, 2878–2890.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Kabir, N.N.; Kazi, J.U. Grb10 is a dual regulator of receptor tyrosine kinase signaling. Mol. Biol. Rep. 2014, 41, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Glidden, E.J.; Gray, L.G.; Vemuru, S.; Li, D.; Harris, T.E.; Mayo, M.W. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem. 2012, 287, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.; Wu, X.; Mao, Z.; Khuri, F.R.; Sun, S.Y. Rictor Undergoes Glycogen Synthase Kinase 3 (GSK3)-dependent, FBXW7-mediated Ubiquitination and Proteasomal Degradation. J. Biol. Chem. 2015, 290, 14120–14129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018, 39, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Shaikenov, T.; Peterson, T.R.; Aimbetov, R.; Bissenbaev, A.K.; Lee, S.W.; Wu, J.; Lin, H.K.; Sarbassov dos, D. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci. Signal. 2011, 4, ra10. [Google Scholar] [CrossRef]
- Julien, L.A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Kwon, B.; Lemere, C.A.; de la Monte, S.; Itamura, K.; Ha, A.Y.; Querfurth, H.W. mTORC2 (Rictor) in Alzheimer’s Disease and Reversal of Amyloid-beta Expression-Induced Insulin Resistance and Toxicity in Rat Primary Cortical Neurons. J. Alzheimer’s Dis. 2017, 56, 1015–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wu, P.; Wang, H.; Zhu, L.; Zhao, W.; Lu, Y. SIN1 promotes the proliferation and migration of breast cancer cells by Akt activation. Biosci. Rep. 2016, 36, e00424. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, X.; Yang, H.; Chang, R.; Kong, C.; Yang, L. SIN1 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition. Cancer 2013, 119, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Gong, L.; Chen, M.; Zhang, Y.; Yuan, H.; Qin, J.; Gao, D. CUL5-SOCS6 complex regulates mTORC2 function by targeting Sin1 for degradation. Cell Discov. 2019, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, M.; Mott, H.R.; Owen, D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem. Soc. Trans. 2020, 48, 1397–1417. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E. Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, N.R.; Kriplani, N.; Hermida, M.A.; Alvarez-Garcia, V.; Wise, H.M. The PTEN protein: Cellular localization and post-translational regulation. Biochem. Soc. Trans. 2016, 44, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, X. Post-translational regulation of PTEN. Oncogene 2008, 27, 5454–5463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Andres, D.A. mTORC2 is required for rit-mediated oxidative stress resistance. PLoS ONE 2014, 9, e115602. [Google Scholar] [CrossRef]
- Kovalski, J.R.; Bhaduri, A.; Zehnder, A.M.; Neela, P.H.; Che, Y.; Wozniak, G.G.; Khavari, P.A. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell 2019, 73, 830–844.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484. [Google Scholar] [CrossRef]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef] [Green Version]
- Khanna, A.; Lotfi, P.; Chavan, A.J.; Montano, N.M.; Bolourani, P.; Weeks, G.; Shen, Z.; Briggs, S.P.; Pots, H.; Van Haastert, P.J.; et al. The small GTPases Ras and Rap1 bind to and control TORC2 activity. Sci. Rep. 2016, 6, 25823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.H.; Jo, U.; Kohrman, A.; Rezaeian, A.H.; Chou, P.C.; Logothetis, C.; Lin, H.K. Posttranslational regulation of Akt in human cancer. Cell Biosci. 2014, 4, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Dai, X.; Laurent, B.; Zheng, N.; Gan, W.; Zhang, J.; Guo, A.; Yuan, M.; Liu, P.; Asara, J.M.; et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019, 21, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Long, J.; Gao, Y.; Zhang, W.; Han, F.; Xu, C.; Sun, L.; Yang, S.C.; Lan, J.; Hou, Z.; et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019, 21, 214–225. [Google Scholar] [CrossRef]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Y.; Zhang, H.; Gao, Y.; Huang, C.; Zhou, A.; Zhou, Y.; Li, Y. Sequential posttranslational modifications regulate PKC degradation. Mol. Biol. Cell 2016, 27, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Robles-Flores, M.; Melendez, L.; Garcia, W.; Mendoza-Hernandez, G.; Lam, T.T.; Castaneda-Patlan, C.; Gonzalez-Aguilar, H. Posttranslational modifications on protein kinase c isozymes. Effects of epinephrine and phorbol esters. Biochim. Biophys. Acta 2008, 1783, 695–712. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A. SGK1: The Dark Side of PI3K Signaling. Curr. Top. Dev. Biol. 2017, 123, 49–71. [Google Scholar]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiang, Y. mTOR Inhibitors at a Glance. Mol. Cell. Pharmacol. 2015, 7, 15–20. [Google Scholar]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, C.E.; German, M.S.; Yang, K.; Wang, J.; VanBrocklin, H.; Regan, M.; Shokat, K.M.; Ducker, G.S.; Kim, G.E.; Hann, B.; et al. A Patient-derived Xenograft Model of Pancreatic Neuroendocrine Tumors Identifies Sapanisertib as a Possible New Treatment for Everolimus-resistant Tumors. Mol. Cancer Ther. 2018, 17, 2702–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Jordan, N.J.; Dutkowski, C.M.; Barrow, D.; Mottram, H.J.; Hutcheson, I.R.; Nicholson, R.I.; Guichard, S.M.; Gee, J.M. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014, 16, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.S.; Findlay, G.M.; Lamb, R.F. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem. Sci. 2005, 30, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Formisano, L.; Napolitano, F.; Rosa, R.; D’Amato, V.; Servetto, A.; Marciano, R.; De Placido, P.; Bianco, C.; Bianco, R. Mechanisms of resistance to mTOR inhibitors. Crit. Rev. Oncol. Hematol. 2020, 147, 102886. [Google Scholar] [CrossRef] [PubMed]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazio, N.; Buzzoni, R.; Baudin, E.; Antonuzzo, L.; Hubner, R.A.; Lahner, H.; WW, D.E.H.; Raderer, M.; Teule, A.; Capdevila, J.; et al. A Phase II Study of BEZ235 in Patients with Everolimus-resistant, Advanced Pancreatic Neuroendocrine Tumours. Anticancer Res. 2016, 36, 713–719. [Google Scholar] [PubMed]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klumpen, H.J.; Capdevila, J.; et al. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naive Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2018, 23, 766.e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III PIK3CA-Mutant ER-Positive and HER2-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerji, U.; Dean, E.J.; Perez-Fidalgo, J.A.; Batist, G.; Bedard, P.L.; You, B.; Westin, S.N.; Kabos, P.; Garrett, M.D.; Tall, M.; et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer Res. 2018, 24, 2050–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; White, S.A.; Hu, K. Role of p90RSK in Kidney and Other Diseases. Int. J. Mol. Sci. 2019, 20, 972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Trans. Sci. 2012, 106, 145–169. [Google Scholar]
- Kumar, A.; Singh, U.K.; Kini, S.G.; Garg, V.; Agrawal, S.; Tomar, P.K.; Pathak, P.; Chaudhary, A.; Gupta, P.; Malik, A. JNK pathway signaling: A novel and smarter therapeutic targets for various biological diseases. Future Med. Chem. 2015, 7, 2065–2086. [Google Scholar] [CrossRef] [PubMed]
- Arrouchi, H.; Lakhlili, W.; Ibrahimi, A. A review on PIM kinases in tumors. Bioinformation 2019, 15, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Song, M.; Kundu, J.K.; Lee, M.H.; Liu, Z.Z. PIM Kinase as an Executional Target in Cancer. J. Cancer Prev. 2018, 23, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015, 5, e325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, S.W.; Sohn, J.H.; Kim, D.H.; Choi, Y.J.; Park, Y.L.; Kim, K.; Cho, Y.H.; Pyo, J.S.; Kim, J.H. Overexpressions of Cyclin B1, cdc2, p16 and p53 in human breast cancer: The clinicopathologic correlations and prognostic implications. Yonsei Med. J. 2011, 52, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, J.A.; Cook, S.J. Targeting IKKbeta in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKbeta Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yan, L.; Liao, N.; Wu, W.Q.; Shi, J.L. A Review of ULK1-Mediated Autophagy in Drug Resistance of Cancer. Cancers 2020, 12, 352. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Goedert, M. GSK3 inhibitors: Development and therapeutic potential. Nat. Rev. Drug Discov. 2004, 3, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.Y.; Eom, J.I.; Jang, J.E.; Jeung, H.K.; Chung, H.; Kim, J.S.; Cheong, J.W.; Min, Y.H. ULK1 inhibition as a targeted therapeutic strategy for FLT3-ITD-mutated acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2020, 39, 85. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. IScience 2018, 8, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Nihira, N.T.; Inuzuka, H.; Wei, W. Physiological functions of FBW7 in cancer and metabolism. Cell Signal. 2018, 46, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. (Lond.) 2018, 38, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, M.; Marusawa, H.; Wang, H.Q.; Iwai, A.; Ikeuchi, K.; Imai, Y.; Kataoka, A.; Nukina, N.; Takahashi, R.; Chiba, T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 2008, 27, 6002–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Moten, A.; Peng, D.; Hsu, C.C.; Pan, B.S.; Manne, R.; Li, H.Y.; Lin, H.K. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin. Cancer Biol. 2020, 67, 16–33. [Google Scholar] [CrossRef]
- Chan, C.H.; Morrow, J.K.; Li, C.F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Moutouh-de Parseval, L.; et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Grigoryan, A.V.; Li, Y.; Hao, B.; Pagano, M.; Cardozo, T.J. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol. 2012, 19, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Imaizumi, K.; Kaneko, M. The Role of Tissue-Specific Ubiquitin Ligases, RNF183, RNF186, RNF182 and RNF152, in Disease and Biological Function. Int. J. Mol. Sci. 2020, 21, 3921. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.I.; Zhu, S.; Gokhale, S.; Jin, J.; Xie, P. TRAF molecules in inflammation and inflammatory diseases. Curr. Pharmacol. Rep. 2018, 4, 64–90. [Google Scholar] [CrossRef]
- Zhu, S.; Jin, J.; Gokhale, S.; Lu, A.M.; Shan, H.; Feng, J.; Xie, P. Genetic Alterations of TRAF Proteins in Human Cancers. Front. Immunol. 2018, 9, 2111. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.; Hospenthal, M.K.; Geurink, P.P.; Elliott, P.R.; Akutsu, M.; Arnaudo, N.; Ekkebus, R.; Kulathu, Y.; Wauer, T.; El Oualid, F.; et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 2013, 154, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Wang, H.; Xiao, Y.; Jin, J.; Chang, J.H.; Zou, Q.; Xie, X.; Cheng, X.; Sun, S.C. Otud7b facilitates T cell activation and inflammatory responses by regulating Zap70 ubiquitination. J. Exp. Med. 2016, 213, 399–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Brittain, G.C.; Chang, J.H.; Puebla-Osorio, N.; Jin, J.; Zal, A.; Xiao, Y.; Cheng, X.; Chang, M.; Fu, Y.X.; et al. OTUD7B controls non-canonical NF-kappaB activation through deubiquitination of TRAF3. Nature 2013, 494, 371–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareja, F.; Ferraro, D.A.; Rubin, C.; Cohen-Dvashi, H.; Zhang, F.; Aulmann, S.; Ben-Chetrit, N.; Pines, G.; Navon, R.; Crosetto, N.; et al. Deubiquitination of EGFR by Cezanne-1 contributes to cancer progression. Oncogene 2012, 31, 4599–4608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.D.; Shen, Y.; Qiao, S.; Liu, W.W.; Zheng, L.; Wang, Y.N.; Cui, N.; Wang, Y.F.; Zhao, S.; Shi, J.H. Upregulation of OTUD7B (Cezanne) Promotes Tumor Progression via AKT/VEGF Pathway in Lung Squamous Carcinoma and Adenocarcinoma. Front. Oncol. 2019, 9, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setsuie, R.; Wada, K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem. Int. 2007, 51, 105–111. [Google Scholar] [CrossRef]
- Wu, H.Q.; Baker, D.; Ovaa, H. Small molecules that target the ubiquitin system. Biochem. Soc. Trans. 2020, 48, 479–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottis, P.; Crews, C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Arvinas’s PROTACs pass first safety and PK analysis. Nat. Rev. Drug Discov. 2019, 18, 895. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, P.; Wei, W. mTOR signaling in tumorigenesis. Biochim. Biophys. Acta 2014, 1846, 638–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Target | PTM | Sites | Enzyme | Influence on mTOR | References |
|---|---|---|---|---|---|
| mTOR | phosphorylation | S2448 | AktS6K1 | activation | [43,44,45,46] |
| mTOR | phosphorylation | S2446 | AMPK | inhibition | [47] |
| mTOR | phosphorylation | S1261 | PI3K/TSC | activation | [48] |
| mTOR | phosphorylation | S2481 | mTOR | activation | [49,50,51] |
| mTOR | phosphorylation | S2159/T2164 | unknown | activation | [52] |
| mTOR | ubiquitination | unknown | FBXW7 | inhibition | [55,56] |
| mTOR | ubiquitination | K2066/K2306 | Parkin | activation | [57] |
| mTOR | malonylation | K1218 | unknown | inhibition | [58] |
| mLST8 | ubiquitination | K305/K313 | TRAF2 | inhibition | [62] |
| mLST8 | deubiquitination | K305/K313 | OTUD7B | activation | [62] |
| DEPTOR | ubiquitination | unknown | SCFβ-TRCP | activation | [64,65,66] |
| DEPTOR | phosphorylation | multiple S/T | p38γ or p38δ | activation | [67] |
| Raptor | phosphorylation | S696/T706/S863 | JNK | activation | [73,74] |
| Raptor | phosphorylation | multiple S | cdc2 | activation | [75,76] |
| Raptor | phosphorylation | S719/S721/S722 | RSKs | activation | [77] |
| Raptor | phosphorylation | S722/S792 | AMPK | inhibition | [78] |
| Raptor | phosphorylation | S606 | LATS | inhibition | [79] |
| Raptor | ubiquitination | unknown | DDB1/CUL4 | activation | [80] |
| Raptor | ubiquitination | unknown | UCH-L1 | inhibition | [81] |
| Raptor | acetylation | unknown | EP300 | activation | [82] |
| PRAS40 | phosphorylation | T246 | Akt or PIM1 | activation | [86] |
| PRAS40 | phosphorylation | S183/S221 | mTORC1 | activation | [86] |
| PRAS40 | phosphorylation | S202/S203 | PKM2 | activation | [86] |
| TSC | phosphorylation | S939/T1462 | Akt | inhibition | [25,26] |
| TSC | phosphorylation | S664 | ERK | inhibition | [96,97] |
| TSC | phosphorylation | S1798 | RSK1 | activation | [98] |
| TSC2 | phosphorylation | S1387 | AMPK | inhibition | [99] |
| TSC1 | phosphorylation | S487/S511 | IKK β | activation | [100] |
| TSC1/2 | ubiquitination | unknown | TRIM31 | activation | [101] |
| TSC | ubiquitination | unknown | Pam | activation | [102] |
| TSC2 | ubiquitination | unknown | E6AP | activation | [103] |
| TSC2 | ubiquitination | unknown | DDB1/ROC1 | activation | [104] |
| Rheb | farnesylation | unknown | unknown | activation | [105,106] |
| Rheb | phosphorylation | S130 | PARK | inhibition | [107] |
| Rheb | ubiquitination | K8 | RNF152 | inhibition | [108] |
| Rheb | deubiquitination | unknown | ATXN3 | inhibition | [109] |
| Rag A | ubiquitination | multiple lysine | RNF152 | inhibition | [114] |
| Rag A | ubiquitination | unknown | Skp2 | inhibition | [115] |
| Rag C | phosphorylation | S2/S21/T394 | mTORC1 | activation | [116] |
| p18 | myristoylation | unknown | unknown | activation | [117] |
| p18 | palmitoylation | unknown | unknown | activation | [117] |
| p18 | ubiquitination | unknown | UBE3A | inhibition | [118] |
| C7orf59 | phosphorylation | S67 | PKA | unknown | [121] |
| DEPDC5 | ubiquitination | multiple lysine | KLHL22 | activation | [122] |
| DEPDC5 | phosphorylation | S1530 | Pim/Akt | activation | [123] |
| FLCN | phosphorylation | S62/S73 | mTORC1 | unknown | [128] |
| FLCN | phosphorylation | S406/S537/S542 | ULK1 | unknown | [129] |
| FLCN | phosphorylation | S302 | unknown | unknown | [130] |
| FLCN | ubiquitination | K206/K559 | unknown | unknown | [131,132] |
| Sestrin2 | phosphorylation | S73/S254 | ULK1 | inhibition | [134,135] |
| Sestrin2 | ubiquitination | K13 | RNF186 | inhibition | [136] |
| S6K1 | phosphorylation | T389 | mTORC1 | activation | [141] |
| S6K1 | phosphorylation | T229 | PDK1 | activation | [142,143] |
| S6K1 | acetylation | C-terminal | p300/PCAF | inhibition | [145] |
| S6K1 | ubiquitination | unknown | ROC1 | inhibition | [146] |
| ULK1 | phosphorylation | S317/S377 | AMPK | activation | [147] |
| ULK1 | phosphorylation | S757 | mTORC1 | inhibition | [147] |
| ULK1 | O-GlcNAcylation | T754 | OGT | activation | [148] |
| ULK1 | acetylation | K162/K606 | TIP60 | activation | [149] |
| ULK1 | ubiquitination | unknown | TRAF6 | inhibition | [150] |
| Grb10 | phosphorylation | S501/S503 | mTORC1 | inhibition | [128] |
| Rictor | acetylation | multiple lysine | p300 | activation | [152] |
| Rictor | phosphorylation | T1695 | GSK3 | inhibition | [153] |
| Rictor | phosphorylation | S1235 | Akt | inhibition | [155] |
| Rictor | phosphorylation | T1135 | S6K1 | activation | [156] |
| Sin1 | phosphorylation | T86/T398 | S6K1 or Akt | inhibition | [161] |
| Sin1 | phosphorylation | T86 | Akt | activation | [162] |
| Sin1 | ubiquitination | multiple lysine | CUL5-SOC6 | inhibition | [163] |
| Akt | phosphorylation | S477/S479 | cyclin A/CDK2 | activation | [176] |
| Akt | ubiquitination | unknown | TRAF6, Skp2 | activation | [177,178] |
| Akt | methylation | K64/K140/K42 | SETDB1 | activation | [179,180] |
| Akt | hydroxylation | P125/P313 | EglN1 | inhibition | [181] |
| PKCα | phosphorylation | unknown | mTORC2 | activation | [60,183,184] |
| SGK1 | phosphorylation | unknown | mTORC2 | activation | [187] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, S.; Liu, L.; Gan, W. The Roles of Post-Translational Modifications on mTOR Signaling. Int. J. Mol. Sci. 2021, 22, 1784. https://doi.org/10.3390/ijms22041784
Yin S, Liu L, Gan W. The Roles of Post-Translational Modifications on mTOR Signaling. International Journal of Molecular Sciences. 2021; 22(4):1784. https://doi.org/10.3390/ijms22041784
Chicago/Turabian StyleYin, Shasha, Liu Liu, and Wenjian Gan. 2021. "The Roles of Post-Translational Modifications on mTOR Signaling" International Journal of Molecular Sciences 22, no. 4: 1784. https://doi.org/10.3390/ijms22041784
APA StyleYin, S., Liu, L., & Gan, W. (2021). The Roles of Post-Translational Modifications on mTOR Signaling. International Journal of Molecular Sciences, 22(4), 1784. https://doi.org/10.3390/ijms22041784