
Remodeling of Bone Marrow Niches and Roles of Exosomes in Leukemia

 ,
,  ,
,  and
and 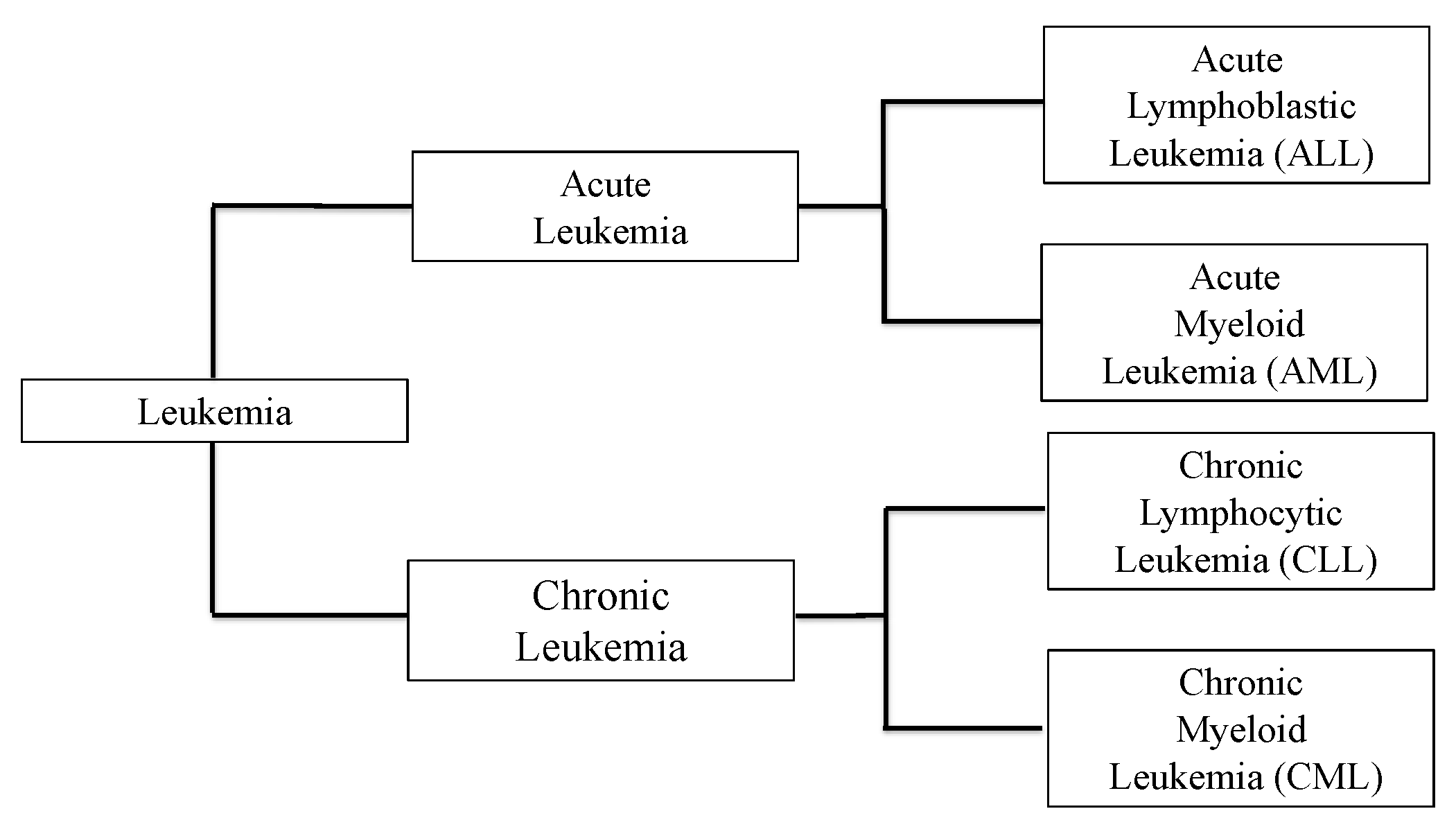{kind=link}
{kind=link}
Abstract
1. Introduction
2. Leukemic Bone Marrow Niche
2.1. The Bone Marrow Niche as an Accomplice to Leukemia
2.2. Leukemic Bone Marrow Niche and Chemotherapy
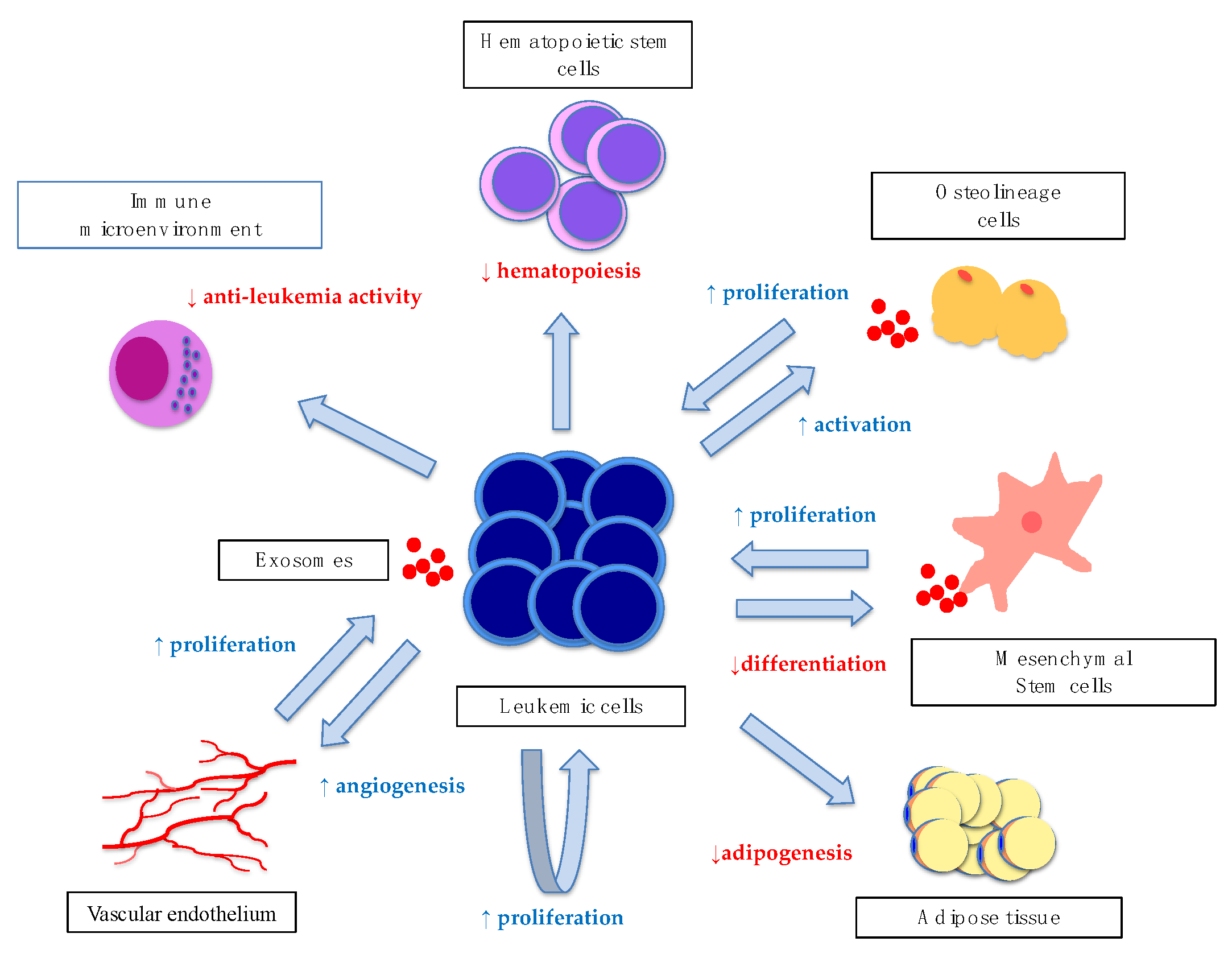
3. Leukemic Exosomes and Bone Marrow Niche Remodeling
3.1. Leukemic Exosomes
3.2. Bone Marrow Remodeling by Leukemic Exosomes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphocytic Leukemia |
| AML | Acute Myeloid Leukemia |
| Bcl2 | B Cell Lymphoma 2 |
| BCR-ABL | Breakpoint Cluster Region - Abelson |
| BM | Bone Marrow |
| BMMSC | Bone Marrow-Derived Mesenchymal Stem Cell |
| BMSC | Bone Marrow Stromal Cell |
| CAR | C-X-C Motif Chemokine Ligand-12-abundant reticular |
| CBX | Carbenoxolone |
| CLL | Chronic Lymphocytic Leukemia |
| CML | Chronic Myeloid Leukemia |
| CNS | Central Nervous System |
| CTLA-4 | Cytotoxic T Lymphocyte-associated Antigen 4 |
| Cx | Connexin |
| CXCL2 | C-X-C Motif Chemokine Ligand 2 |
| CXCL12 | C-X-C Motif Chemokine Ligand-12 |
| DKK-1 | Dickkopf-1 |
| EC | Endothelial Cell |
| EV | Extracellular Vesicle |
| HEV | High Endothelial Venule |
| HIF | Hypoxia Inducible Factor |
| HSC | Hematopoietic Stem Cell |
| HSCT | Hematopoietic Stem Cell Transplantation |
| HSPC | Hematopoietic stem and Progenitor Cell |
| HUVEC | Human Umbilical Vein Endothelial Cell |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| IL-8 | Interleukin-8 |
| IL-15 | Interleukin-15 |
| IL-15Rα | Interleukin-15 Receptor Alpha |
| K3 | Kindlin-3 |
| LFA-1 | Lymphocyte Function-associated Antigen-1 |
| IGF1 | Insulin-Like Growth Factor-1 |
| LSC | Leukemic Stem Cell |
| LT-HSC | Long-Term Hematopoietic Stem Cell |
| MHC | Major Histocompatibility Complex |
| MRD | Minimal Residual Disease |
| MSC | Mesenchymal Stem Cell |
| MAdCAM-1 | Mucosal Addressin Cell Adhesion Molecule-1 |
| miR | Micro RNA |
| mTOR | Mammalian Target of Rapamycin |
| NK | Natural Killer |
| NKGD2 | Natural Killer Group 2, Member D |
| NKX2-3 | NK2 Homeobox 3 |
| Ph+ | Philadelphia Chromosome-Positive |
| ROS | Reactive Oxygen Species |
| RUNX | Runt-related Transcription Factor |
| STC1 | Stanniocalcin 1 |
| TGF-β1 | Transforming Growth Factor–beta 1 |
| TKI | Tyrosine Kinase Inhibitor |
| TNF | Tumor Necrosis Factor |
| VCAM-1 | Vascular Cell Adhesion Molecule-1 |
| VEGF-AA | Vascular Endothelial Growth Factor A-A |
| VLA-4 | Very Late Antigen-4 |
| WHO | World Health Organization |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Gökbuget, N.; Stanze, D.; Beck, J.; Diedrich, H.; Horst, H.A.; Hüttmann, A.; Kobbe, G.; Kreuzer, K.A.; Leimer, L.; Reichle, A.; et al. German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia. Blood 2012, 120, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Dinner, S.; Lee, D.; Liedtke, M. Current therapy and novel agents for relapsed or refractory acute lymphoblastic leukemia. Leuk. Lymphoma 2014, 55, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Reagan, M.R.; Rosen, C.J. Navigating the bone marrow niche: Translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol. 2016, 12, 154–168. [Google Scholar] [CrossRef]
- He, N.; Zhang, L.; Cui, J.; Li, Z. Bone marrow vascular niche: Home for hematopoietic stem cells. Bone Marrow Res. 2014, 128436. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M. Role of microenvironment in resistance to therapy in AML. Curr. Hematol. Malig. Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef]
- Nagasawa, T.; Omatsu, Y.; Sugiyama, T. Control of hematopoietic stem cells by the bone marrow stromal niche: The role of reticular cells. Trends Immunol. 2011, 32, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Van Noorden, C.J.F.; Carraway, H.E.; Maciejewski, J.P.; Molenaar, R.J. Novel therapeutic strategies to target leukemic cells that hijack compartmentalized continuous hematopoietic stem cell niches. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 183–198. [Google Scholar] [CrossRef]
- Pando, A.; Reagan, J.L.; Quesenberry, P.; Fast, L.D. Extracellular vesicles in leukemia. Leuk. Res. 2018, 64, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Kawamoto, E.; Gaowa, A.; Okamoto, T.; Park, E.J. Connexins and integrins in exosomes. Cancers 2019, 11, 106. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes:composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S.N.; Abdel-Mageed, A.B. Tumor-derived exosomes in oncogenic reprogramming and cancer progression. Cell. Mol. Life Sci. 2014, 72, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, T.; Kosaka, N.; Ochiya, T. The roles of extracellular vesicles in cancer biology: Toward the development of novel cancer biomarkers. Proteomics 2014, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, I.; Lamorte, D.; Trino, S.; De Luca, L.; Ambrosino, C.; Zoppoli, P.; Ruggieri, V.; Del Vecchio, L.; Musto, P.; Caivano, A.; et al. Extracellular vesicles: A new prospective in crosstalk between microenvironment and stem cells in hematological malignancies. Stem Cells Int. 2018, 2018, 9863194. [Google Scholar] [CrossRef] [PubMed]
- Ayala, F.; Dewar, R.; Kieran, M.; Kalluri, R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia 2009, 23, 2233–2241. [Google Scholar] [CrossRef]
- Sánchez-Aguilera, A.; Méndez-Ferrer, S. The hematopoietic stem-cell niche in health and leukemia. Cell. Mol. Life Sci. 2017, 74, 579–590. [Google Scholar] [CrossRef]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Dhami, S.P.S.; Kappala, S.S.; Thompson, A.; Szegezdi, E. Three- dimensional ex vivo co-culture models of the leukaemic bone marrow niche for functional drug testing. Drug. Discov. Today 2016, 21, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Goloviznina, N.A.; Kamimae-Lanning, A.N.; David, L.L.; Wilmarth, P.A.; Mori, T.; Chevillet, J.R.; Narla, A.; Roberts, C.T., Jr.; et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 2015, 29, 2285–2295. [Google Scholar] [CrossRef]
- Cogle, C.R.; Goldman, D.C.; Madlambayan, G.J.; Leon, R.P.; Al Masri, A.; Clark, H.A.; Asbaghi, S.A.; Tyner, J.W.; Dunlap, J.; Fan, G.; et al. Functional integration of acute myeloid leukemia into the vascular niche. Leukemia 2014, 28, 1978–1987. [Google Scholar] [CrossRef]
- Hussong, J.W.; Rodgers, G.M.; Shami, P.J. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 2000, 95, 309–313. [Google Scholar] [CrossRef]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef][Green Version]
- Galán-Díez, M.; Kousteni, S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr. Mol. Biol. Rep. 2017, 3, 53–62. [Google Scholar] [CrossRef]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef]
- Chen, S.Y.; Yang, X.; Feng, W.L.; Liao, J.F.; Wang, L.N.; Feng, L.; Lin, Y.M.; Ren, Q.; Zheng, G.G. Organ-specific microenvironment modifies diverse functional and phenotypic characteristics of leukemia-associated macrophages in mouse T cell acute lymphoblastic leukemia. J. Immunol. 2015, 194, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; You, M.J.; Yang, Y.; Hu, D.; Tian, C. The Role of Tumor-Associated Macrophages in Leukemia. Acta Haematol. 2020, 143, 112–117. [Google Scholar] [CrossRef]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Dührsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and heamatopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Koh, Y.J.; Moon, H.R.; Ryoo, H.G.; Cho, C.H.; Kim, I.; Koh, G.Y. Adipose tissue is an extramedullary reservoir for functional hematopoietic stem and progenitor cells. Blood 2010, 115, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.L.; Reid, J.C.; Salci, K.R.; Aslostovar, L.; Benoit, Y.D.; Shapovalova, Z.; Nakanishi, M.; Porras, D.P.; Almakadi, M.; Campbell, C.J.V.; et al. Acute myeloid leukemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat. Cell Biol. 2017, 11, 1336–1347. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Havens, A.M.; Pienta, K.J.; Taichman, R.S. The bone marrow niche: Habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia 2008, 22, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, H.; Li, W.; Deng, Y.; Munegowda, M.A.; Chibbar, R.; Qureshi, M.; Xiang, J. Dendritic cells recruit T cell exosomes via exosomal LFA-1 leading to inhibition of CD8+ CTL responses through downregulation of peptide/MHC class I and Fas ligand-mediated cytotoxicity. J. Immunol. 2010, 185, 5268–5278. [Google Scholar] [CrossRef] [PubMed]
- Kammertoens, T.; Friese, C.; Arina, A.; Idel, C.; Briesemeister, D.; Rothe, M.; Ivanov, A.; Szymborska, A.; Patone, G.; Kunz, S.; et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 2017, 545, 98–102. [Google Scholar] [CrossRef]
- Moser, M.; Legate, K.R.; Zent, R.; Fässler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef]
- Moser, M.; Nieswandt, B.; Ussar, S.; Pozgajova, M.; Fässler, R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 2008, 14, 325–330. [Google Scholar] [CrossRef]
- Moser, M.; Bauer, M.; Schmid, S.; Ruppert, R.; Schmidt, S.; Sixt, M.; Wang, H.V.; Sperandio, M.; Fässler, R. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 2009, 15, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Moretti, F.A.; Moser, M.; Lyck, R.; Abadier, M.; Ruppert, R.; Engelhardt, B.; Fässler, R. Kindlin-3 regulates integrin activation and adhesion reinforcement of effector T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 17005–17010. [Google Scholar] [CrossRef]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef]
- Krenn, P.W.; Koschmieder, S.; Fässler, R. Kindlin-3 loss curbs chronic myeloid leukemia in mice by mobilizing leukemic stem cells from protective bone marrow niches. Proc. Natl. Acad. Sci. USA 2020, 117, 24326–24335. [Google Scholar] [CrossRef]
- Katsumura, K.R.; Ong, I.M.; DeVilbiss, A.W.; Sanalkumar, R.; Bresnick, E.H. GATA factor-dependent positive-feedback circuit in acute myeloid leukemia cells. Cell Rep. 2016, 16, 2428–2441. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Hawkins, E.D.; Akinduro, O.; Ang, H.; De Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77. [Google Scholar] [CrossRef]
- Rauch, P.J.; Ellegast, J.M.; Widmer, C.C.; Fritsch, K.; Goede, J.S.; Valk, P.J.; Löwenberg, B.; Takizawa, H.; Manz, M.G. MPL expression on AML blasts predicts peripheral blood neutropenia and thrombocytopenia. Blood 2016, 128, 2253–2257. [Google Scholar] [CrossRef] [PubMed]
- Waclawiczek, A.; Hamilton, A.; Rouault-Pierre, K.; Abarrategi, A.; Albornoz, M.G.; Miraki-Moud, F.; Bah, N.; Gribben, J.; Fitzgibbon, J.; Taussig, D.; et al. Mesenchymal niche remodeling impairs hematopoiesis via stanniocalcin 1 in acute myeloid leukemia. J. Clin. Invest. 2020, 130, 3038–3050. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef]
- Burger, J.A.; Burkle, A. The CXCR4 chemokine receptor in acute and chronic leukaemia: A marrow homing receptor and potential therapeutic target. Br. J. Haematol. 2007, 137, 288–296. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, Z.; An, J.; Xu, Y. A novel dimeric CXCR4 antagonist synergizes with chemotherapy in acute myeloid leukaemia by mobilizing leukaemic cells from their associated bone marrow niches. Br. J. Haematol. 2019, 187, e11–e15. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017, 19, 214–223. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, Á.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell 2019, 24, 769–784. [Google Scholar] [CrossRef]
- Mesnil, M. Connexins and cancer. Biol. Cell 2002, 94, 493–500. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambon, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef] [PubMed]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Kouzi, F.; Zibara, K.; Bourgeais, J.; Picou, F.; Gallay, N.; Brossaud, J.; Dakik, H.; Roux, B.; Hamard, S.; Le Nail, L.R.; et al. Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene 2020, 39, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, O.; Friedman, D.; Leshkowitz, D.; Goldenberg, D.; Orlovsky, K.; Pencovich, N.; Lotem, J.; Tanay, A.; Groner, Y. Addiction of t (8;21) and inv (16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013, 4, 1131–1143. [Google Scholar] [CrossRef]
- Goyama, S.; Schibler, J.; Cunningham, L.; Zhang, Y.; Rao, Y.; Nishimoto, N.; Nakagawa, M.; Olsson, A.; Wunderlich, M.; Link, K.A.; et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Invest. 2013, 123, 3876–3888. [Google Scholar] [CrossRef] [PubMed]
- Hyde, R.K.; Kamikubo, Y.; Anderson, S.; Kirby, M.; Alemu, L.; Zhao, L.; Liu, P.P. Cbfb/Runx1 repression-independent blockage of differentiation and accumulation of Csf2rb-expressing cells by Cbfb-MYH11. Blood 2010, 115, 1433–1443. [Google Scholar] [CrossRef]
- Morita, K.; Suzuki, K.; Maeda, S.; Matsuo, A.; Mitsuda, Y.; Tokushige, C.; Kashiwazaki, G.; Taniguchi, J.; Maeda, R.; Noura, M.; et al. Genetic regulation of the RUNX transcription factor family has antitumor effects. J. Clin. Invest. 2017, 127, 2815–2828. [Google Scholar] [CrossRef]
- Morita, K.; Tokushige, C.; Maeda, S.; Kiyose, H.; Noura, M.; Iwai, A.; Yamada, M.; Kashiwazaki, G.; Taniguchi, J.; Bando, T.; et al. RUNX transcription factors potentially control E-selectin expression in the bone marrow vascular niche in mice. Blood Adv. 2018, 2, 509–515. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Ohyashiki, J.H.; Umezu, T.; Ohyashiki, K. Exosomes promote bone marrow angiogenesis in hema¬tologic neoplasia: The role of hypoxia. Curr. Opin. Hematol. 2016, 23, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Mineo, M.; Garfield, S.H.; Taverna, S.; Flugy, A.; De Leo, G.; Alessandro, R.; Kohn, E.C. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a Src-dependent fashion. Angiogenesis 2012, 15, 33–45. [Google Scholar] [CrossRef]
- Taverna, S.; Flugy, A.; Saieva, L.; Kohn, E.C.; Santoro, A.; Meraviglia, S.; De Leo, G.; Alessandro, R. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int. J. Cancer 2012, 130, 2033–2043. [Google Scholar] [CrossRef]
- Kinjyo, I.; Bragin, D.; Grattan, R.; Winter, S.S.; Wilson, B.S. Leukemia-derived exosomes and cytokines pave the way for entry into the brain. J. Leukoc. Biol. 2019, 105, 741–753. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The role of cancer-derived microRNAs in cancer immune escape. J. Hematol. Oncol. 2020, 13, 25. [Google Scholar] [CrossRef]
- Hong, C.S.; Muller, L.; Whiteside, T.L.; Boyiadzis, M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front. Immunol. 2014, 5, 160. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef]
- Koolivand, M.; Ansari, M.; Piroozian, F.; Moein, S.; MalekZadeh, K. Alleviating the progression of acute myeloid leukemia (AML) by sulforaphane through controlling miR-155 levels. Mol. Biol. Rep. 2018, 45, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Foroni, C.; Zanaglio, C.; Re, F.; Polverelli, N.; Turra, A.; Morello, E.; Farina, M.; Cattina, F.; Gandolfi, L.; et al. Feasibility of tumor derived exosome enrichment in the onco hematology leukemic model of chronic myeloid leukemia. Int. J. Mol. Med. 2019, 44, 2133–2214. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef]
- Cross, N.C.; White, H.E.; Colomer, D.; Ehrencrona, H.; Foroni, L.; Gottardi, E.; Lange, T.; Lion, T.; Polakova, K.M.; Dulucq, S.; et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia 2015, 29, 999–1003. [Google Scholar] [CrossRef]
- Egan, D.; Radich, J. Monitoring disease burden in chronic myeloid leukemia: Past, present, and future. Am. J. Hematol. 2016, 91, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre stop imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Ross, D.M.; Masszi, T.; Gómez Casares, M.T.; Hellmann, A.; Stentoft, J.; Conneally, E.; Garcia-Gutierrez, V.; Gattermann, N.; le Coutre, P.D.; Martino, B.; et al. Durable treatment-free remission in patients with chronic myeloid leukemia in chronic phase following frontline nilotinib: 96-week update of the ENESTfreedom study. J. Cancer Res. Clin. Oncol. 2018, 144, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Rea, D.; Lipton, J.H. Treatment-free remission with first- and second-generation tyrosine kinase inhibitors. Am. J. Hematol. 2019, 94, 346–357. [Google Scholar] [CrossRef]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The leukemic stem cell niche: Current concepts and therapeutic opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Gao, X.; Wan, Z.; Wei, M.; Dong, Y.; Zhao, Y.; Chen, X.; Li, Z.; Qin, W.; Yang, G.; Liu, L. Chronic myelogenous leukemia cells remodel the bone marrow niche via exosome-mediated transfer of miR-320. Theranostics 2019, 9, 5642–5656. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2019, 32, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Amodeo, V.; Saieva, L.; Russo, A.; Giallombardo, M.; De Leo, G.; Alessandro, R. Exosomal shuttling of miR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol. Cancer 2014, 13, 169. [Google Scholar] [CrossRef]
- Akinduro, O.; Weber, T.S.; Ang, H.; Haltalli, M.L.R.; Ruivo, N.; Duarte, D.; Rashidi, N.M.; Hawkins, E.D.; Duffy, K.R.; Lo Celso, C. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat. Commun. 2018, 9, 519. [Google Scholar] [CrossRef]
- Boyd, A.L.; Campbell, C.J.; Hopkins, C.I.; Fiebig-Comyn, A.; Russell, J.; Ulemek, J.; Foley, R.; Leber, B.; Xenocostas, A.; Collins, T.J.; et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J. Exp. Med. 2014, 211, 1925–1935. [Google Scholar] [CrossRef]
- Abdelhamed, S.; Butler, J.T.; Doron, B.; Halse, A.; Nemecek, E.; Wilmarth, P.A.; Marks, D.L.; Chang, B.H.; Horton, T.; Kurre, P. Extracellular vesicles impose quiescence on residual hematopoietic stem cells in the leukemic niche. EMBO Rep. 2019, 20, 47546. [Google Scholar] [CrossRef] [PubMed]
- Miraki-Moud, F.; Anjos-Afonso, F.; Hodby, K.A.; Griessinger, E.; Rosignoli, G.; Lillington, D.; Jia, L.; Davies, J.K.; Cavenagh, J.; Smith, M.; et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 13576–13581. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, T.; Kawamoto, E.; Gaowa, A.; Park, E.J.; Shimaoka, M. Remodeling of Bone Marrow Niches and Roles of Exosomes in Leukemia. Int. J. Mol. Sci. 2021, 22, 1881. https://doi.org/10.3390/ijms22041881
Yamaguchi T, Kawamoto E, Gaowa A, Park EJ, Shimaoka M. Remodeling of Bone Marrow Niches and Roles of Exosomes in Leukemia. International Journal of Molecular Sciences. 2021; 22(4):1881. https://doi.org/10.3390/ijms22041881
Chicago/Turabian StyleYamaguchi, Takanori, Eiji Kawamoto, Arong Gaowa, Eun Jeong Park, and Motomu Shimaoka. 2021. "Remodeling of Bone Marrow Niches and Roles of Exosomes in Leukemia" International Journal of Molecular Sciences 22, no. 4: 1881. https://doi.org/10.3390/ijms22041881
APA StyleYamaguchi, T., Kawamoto, E., Gaowa, A., Park, E. J., & Shimaoka, M. (2021). Remodeling of Bone Marrow Niches and Roles of Exosomes in Leukemia. International Journal of Molecular Sciences, 22(4), 1881. https://doi.org/10.3390/ijms22041881