
40-Hz Auditory Steady-State Response (ASSR) as a Biomarker of Genetic Defects in the SHANK3 Gene: A Case Report of 15-Year-Old Girl with a Rare Partial SHANK3 Duplication

 , ,
, , 
Abstract
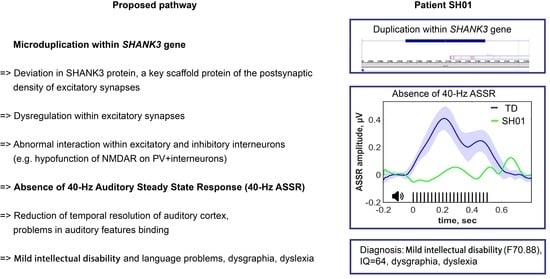
1. Introduction
2. Results
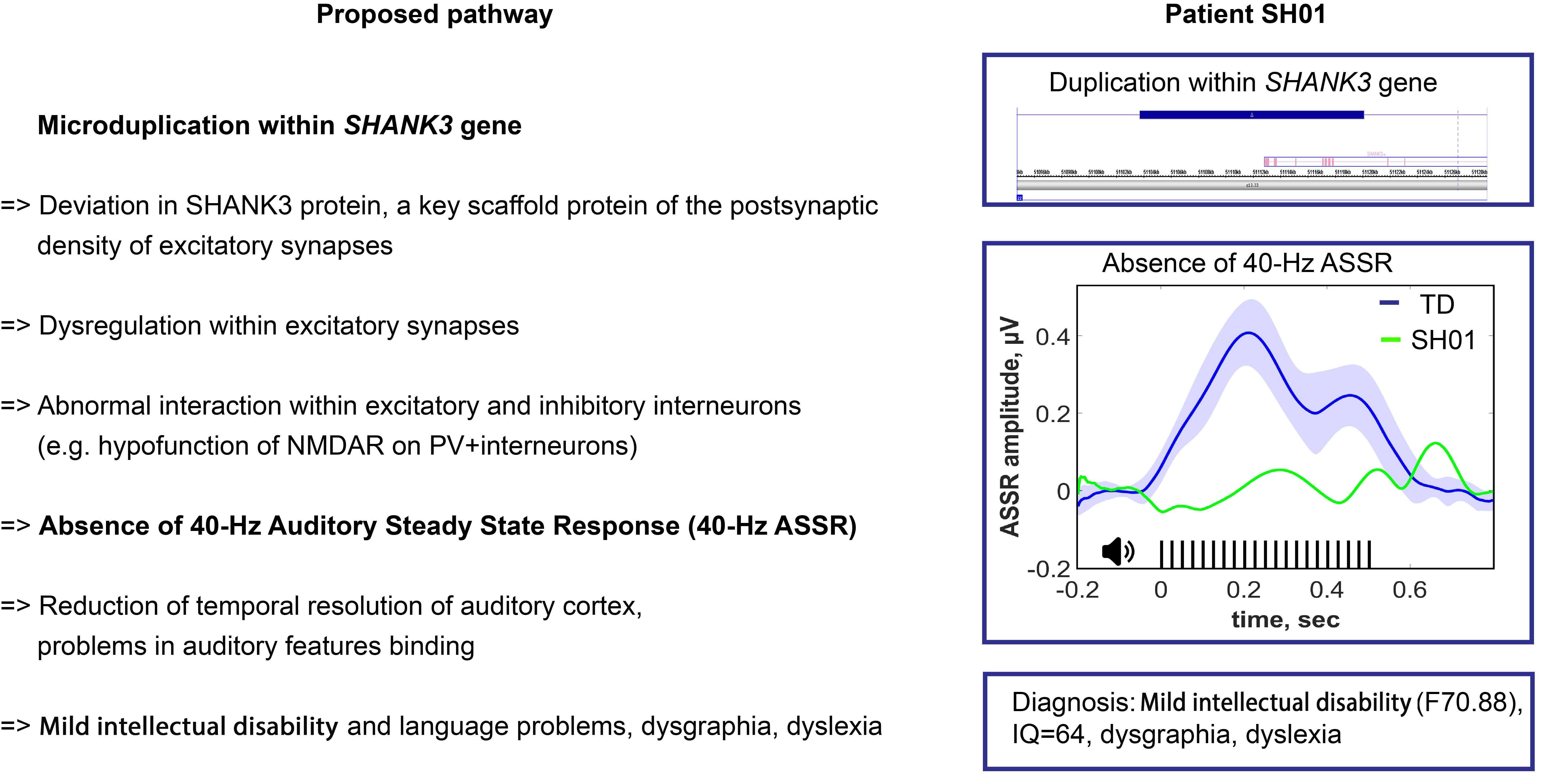
2.1. Genetic Information
2.2. Phenotyping, Clinical Description
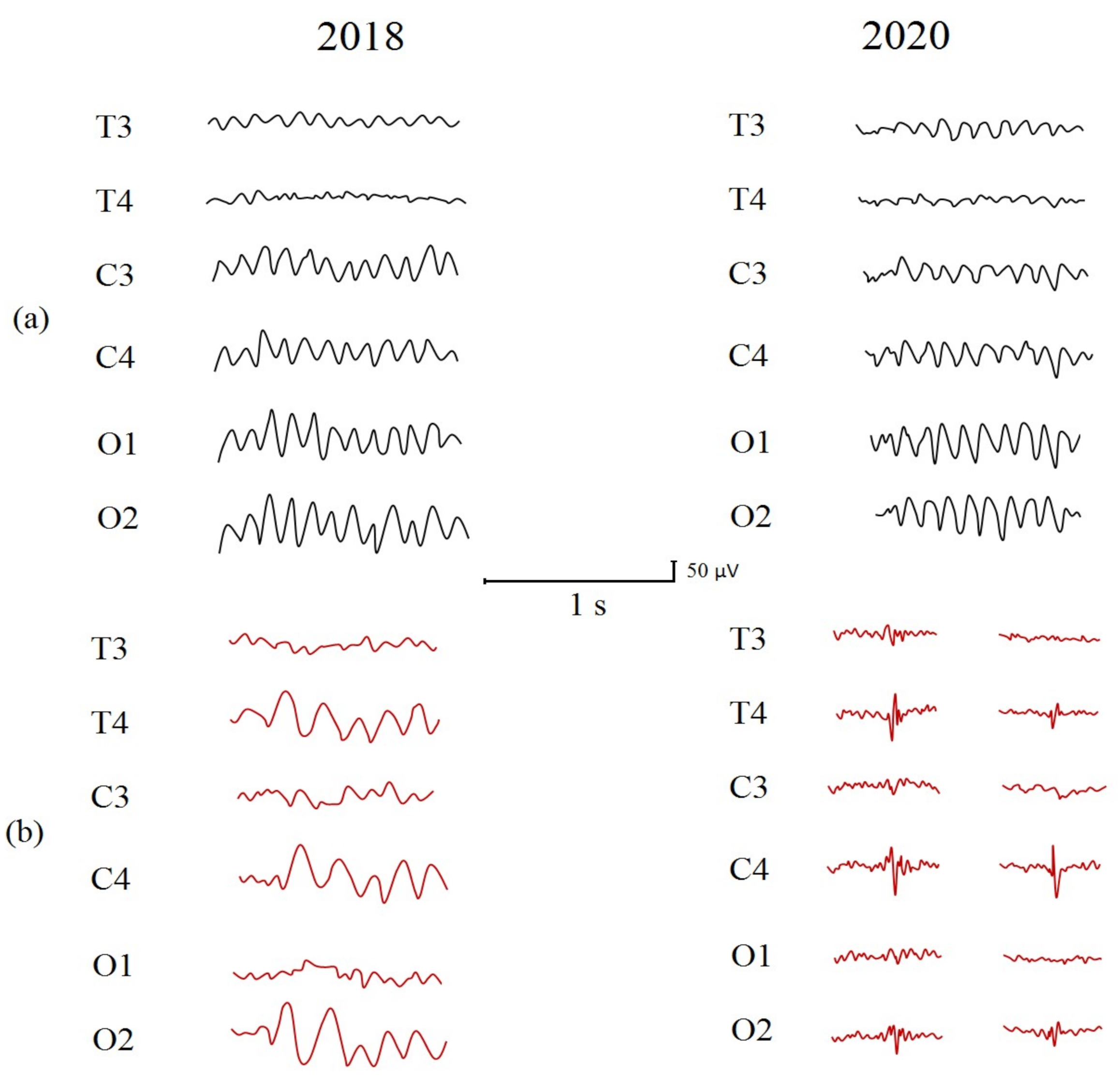
2.3. Clinical EEG
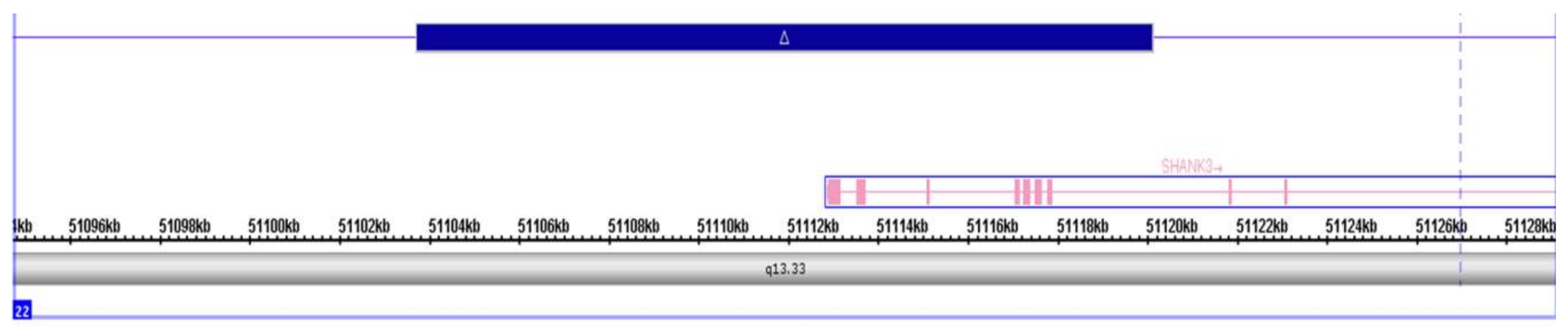
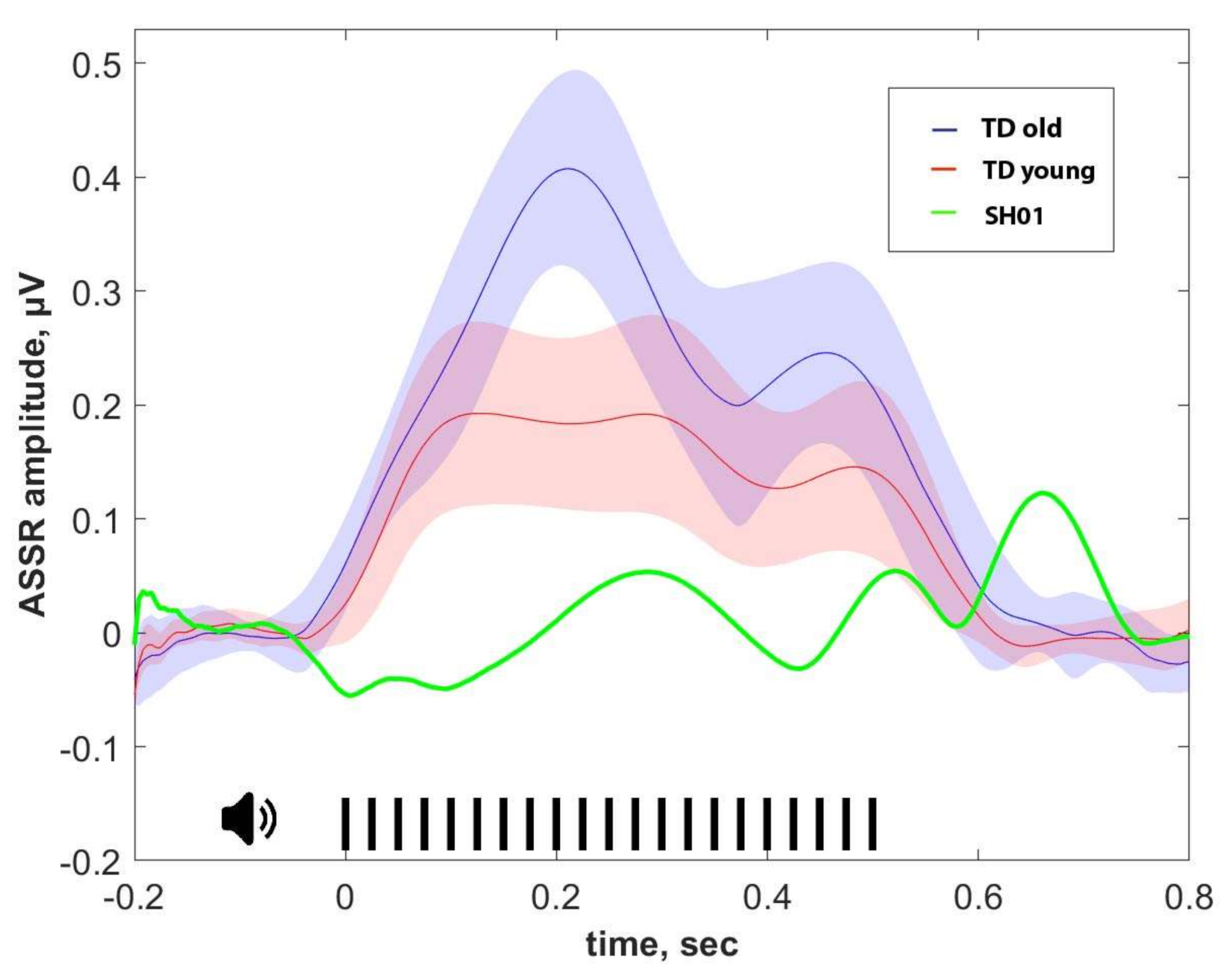
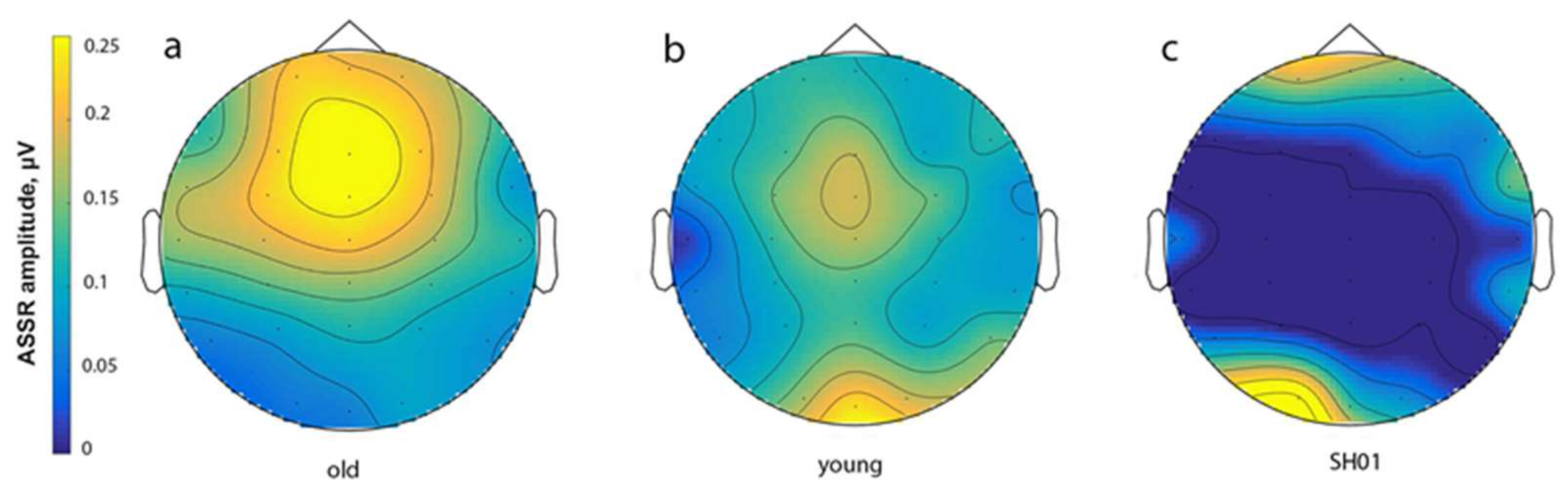
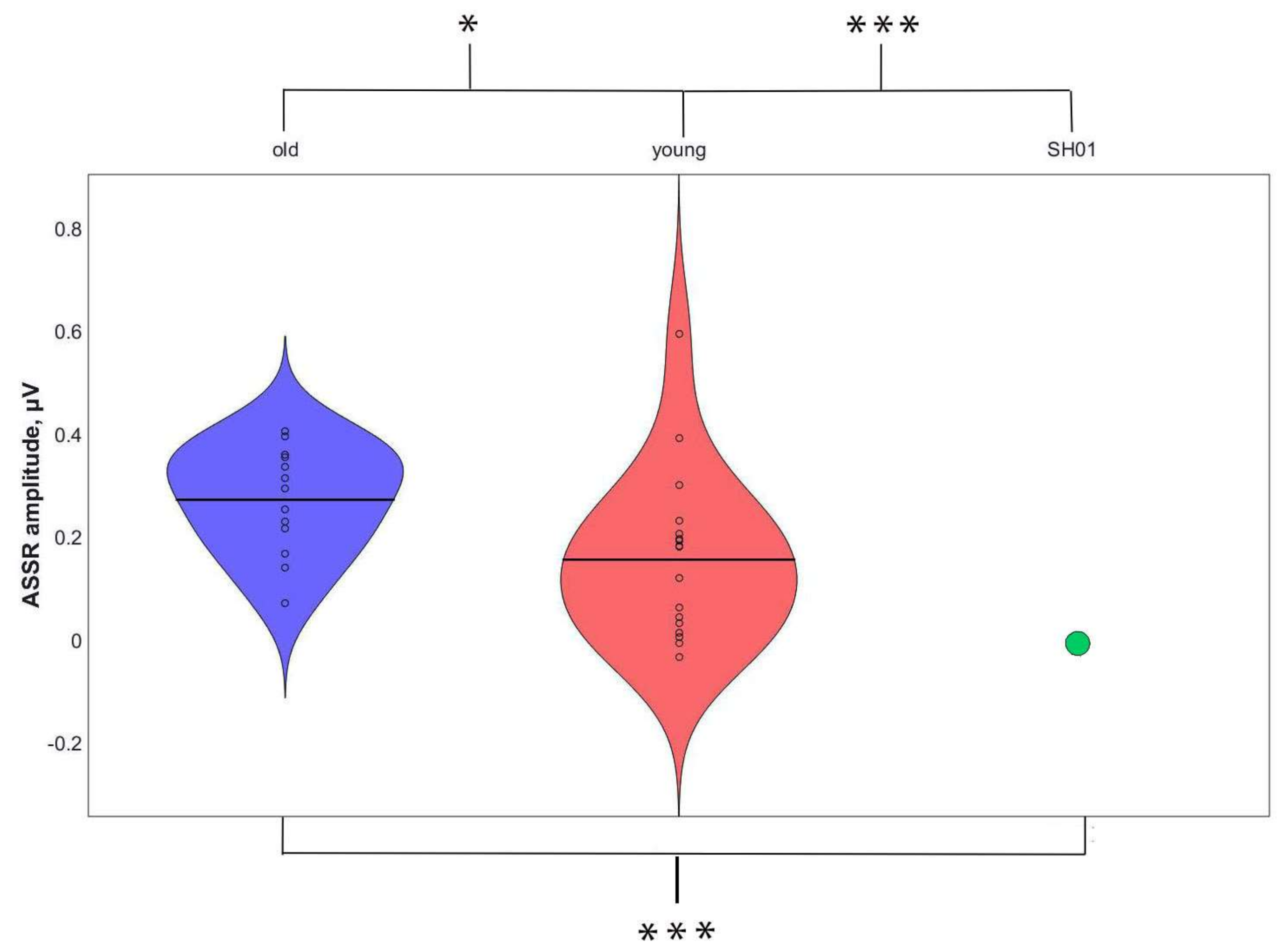
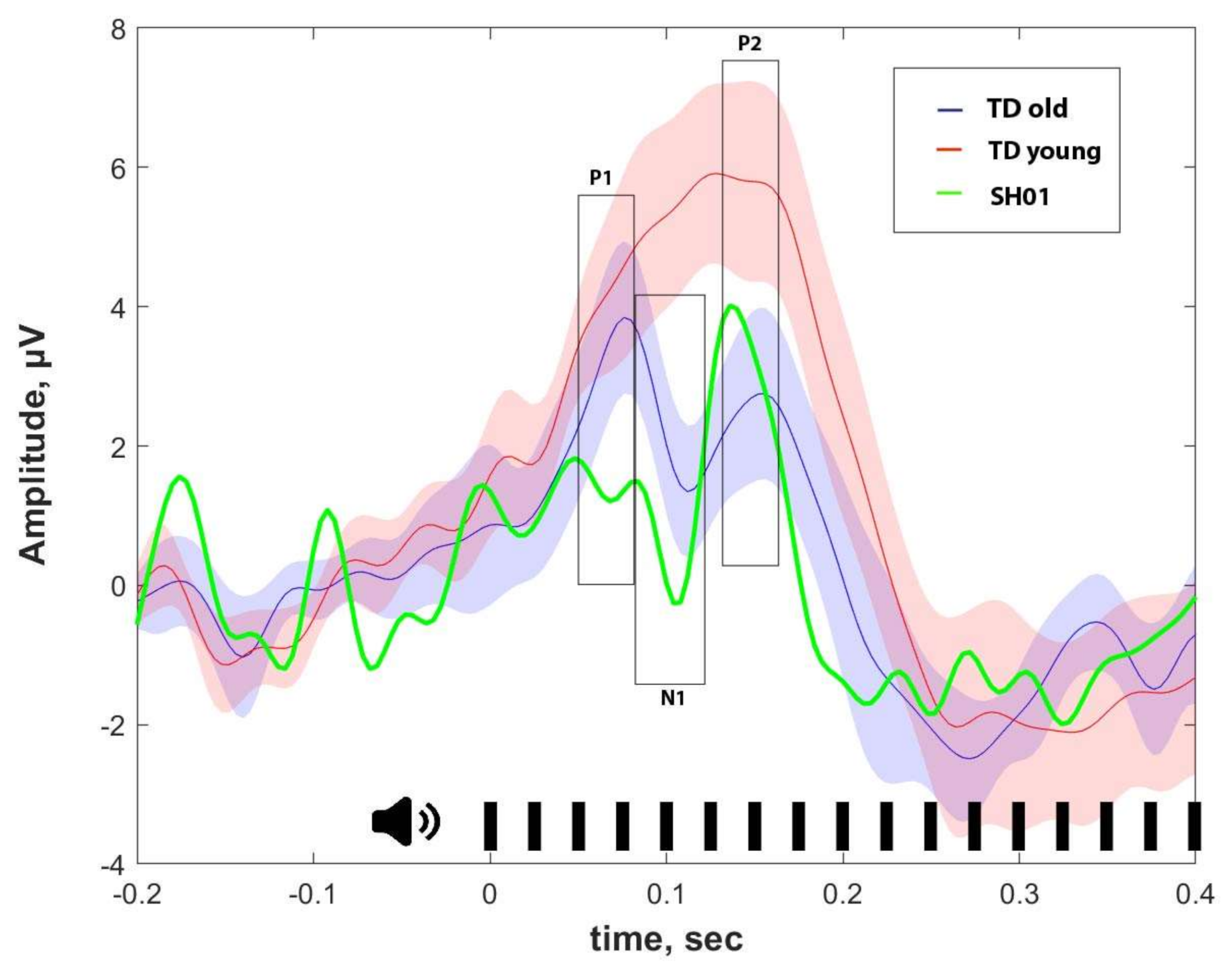
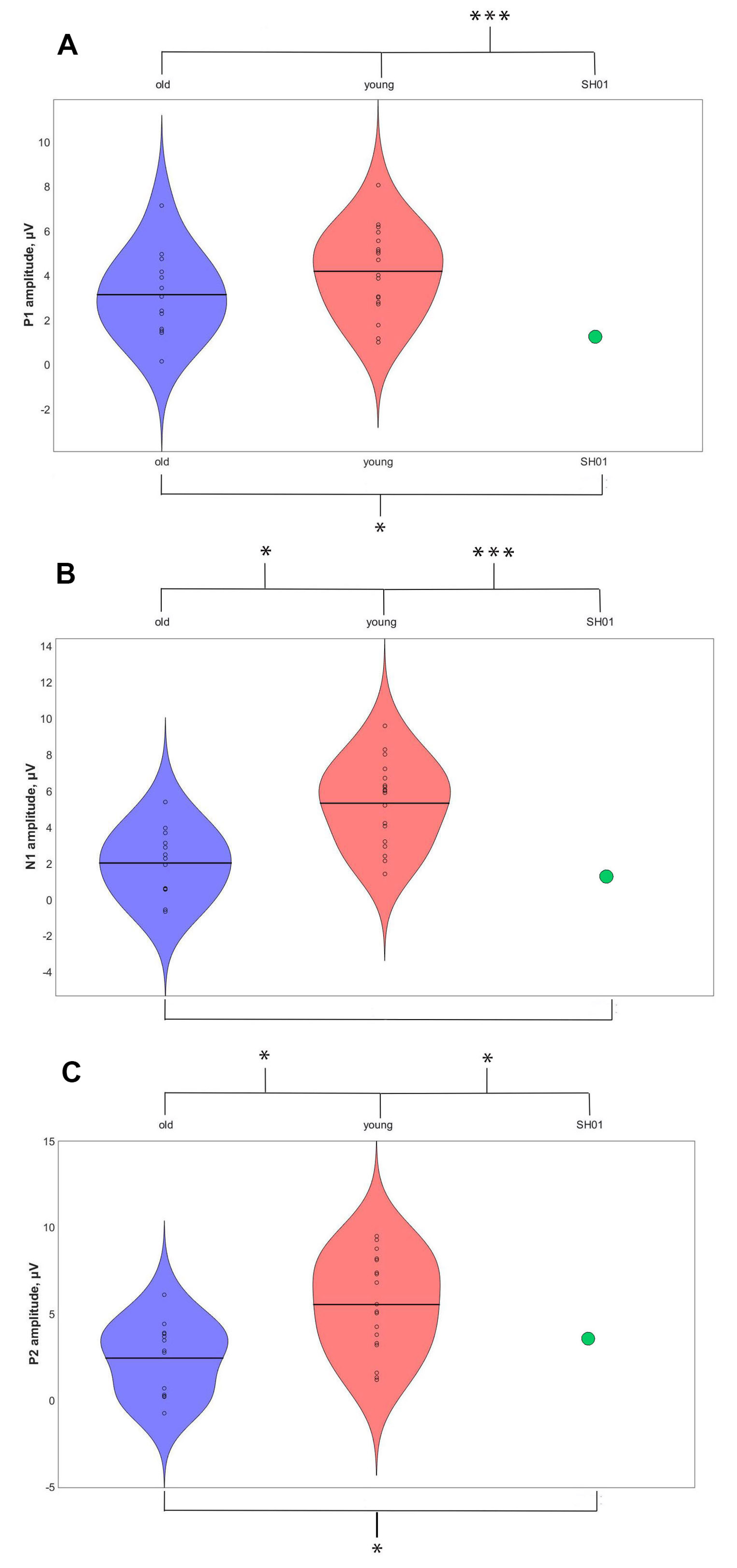
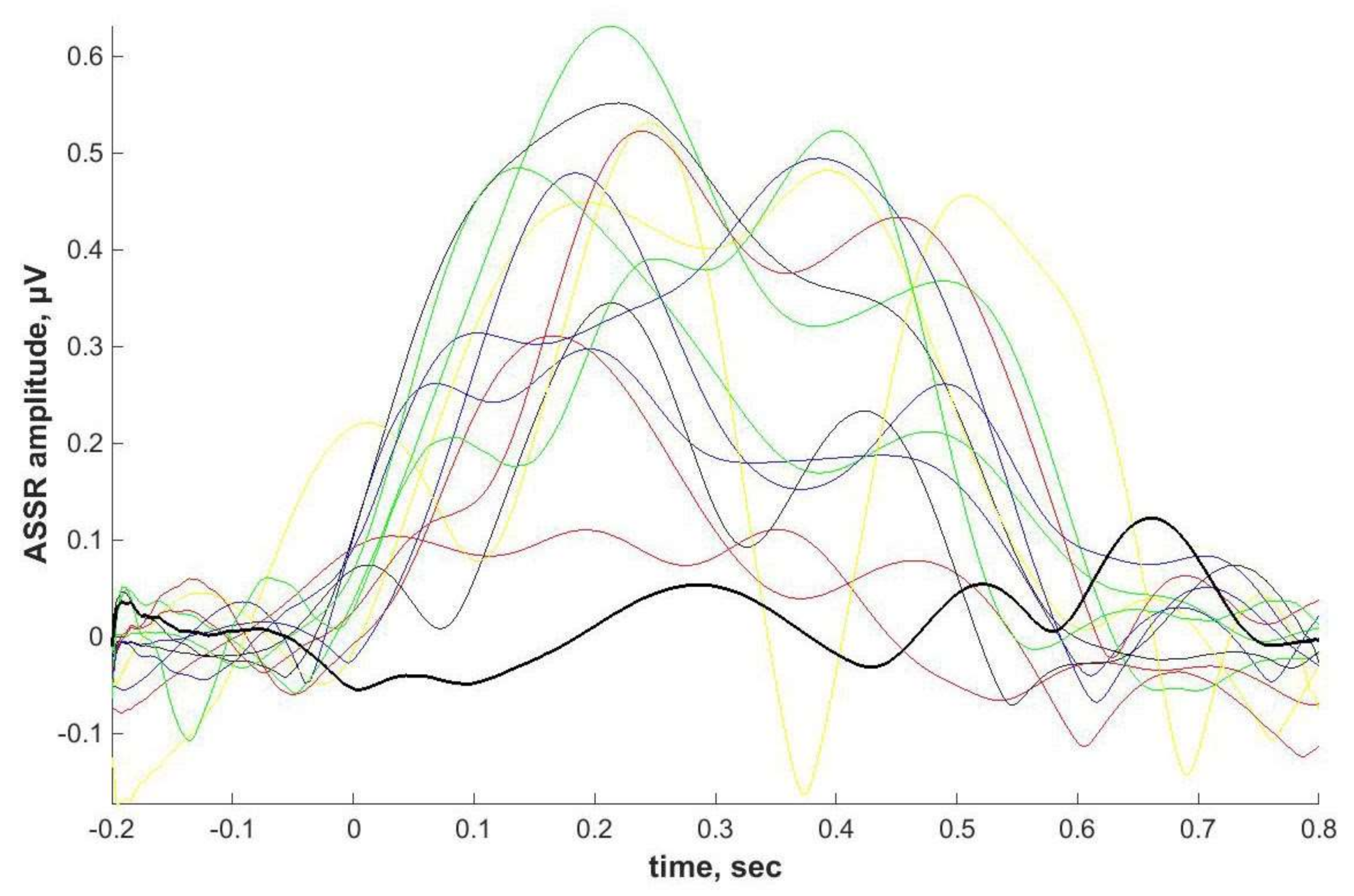
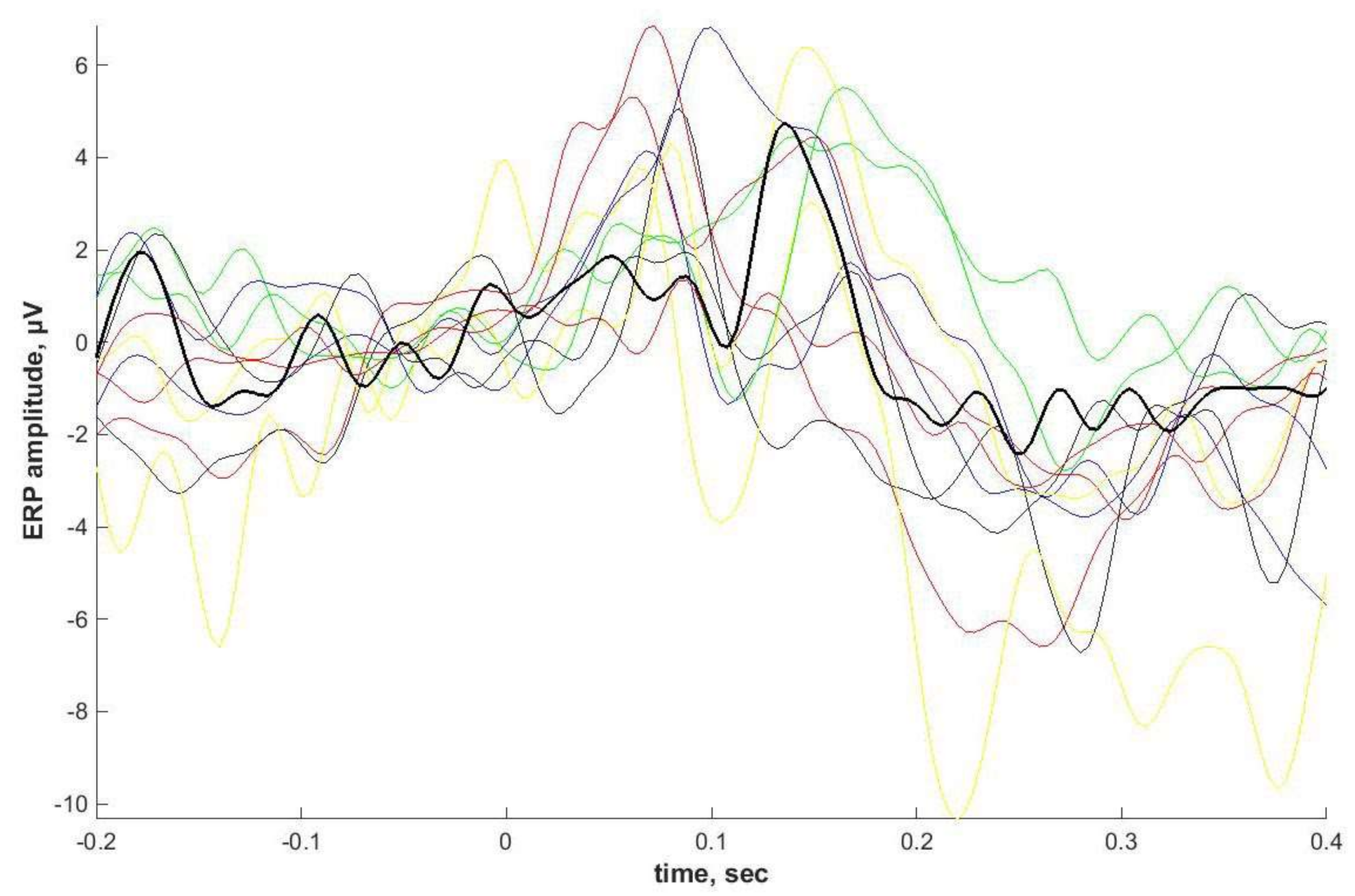
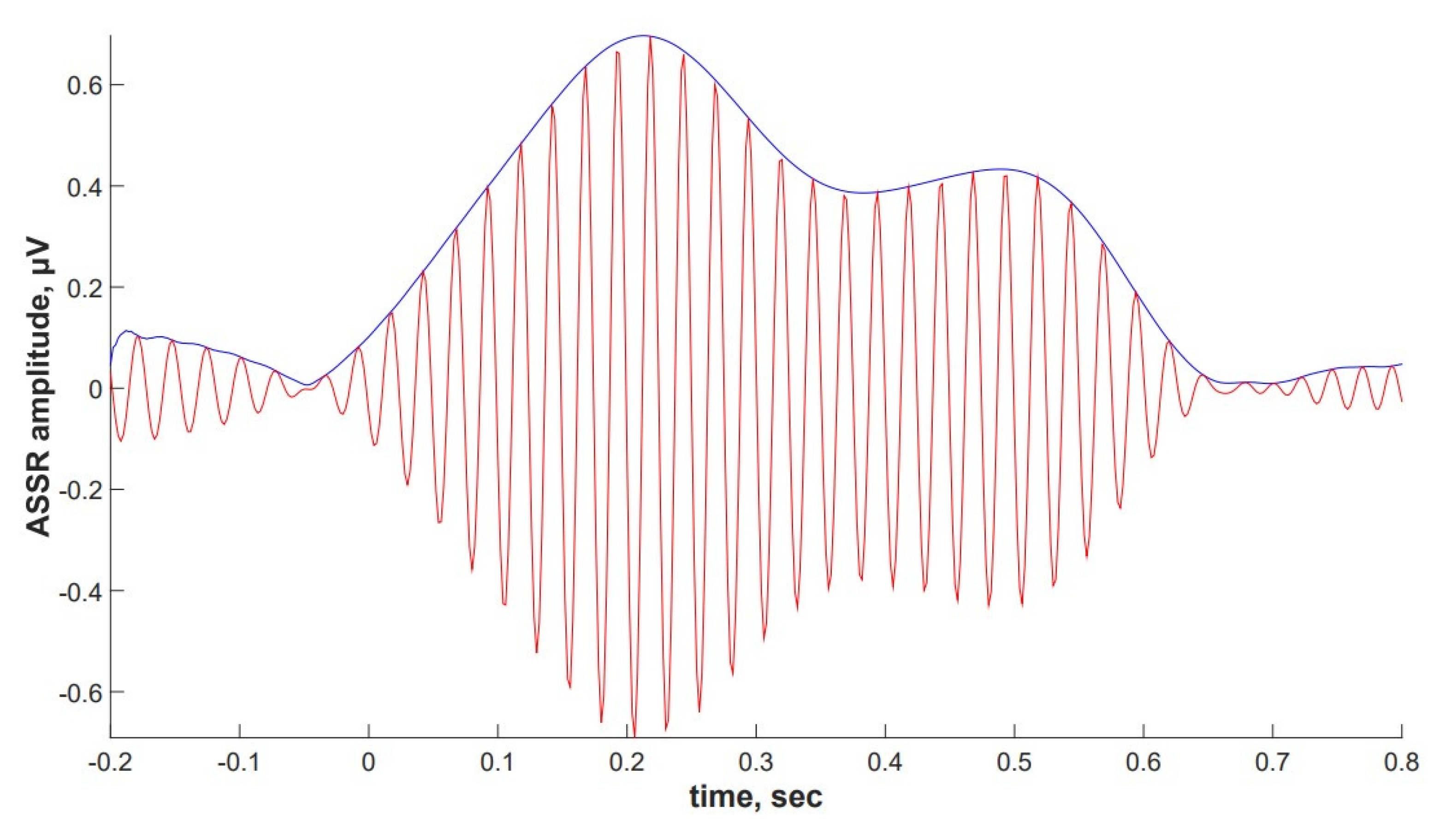
2.4. ASSR/Auditory ERP
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. EEG Recording
4.3. Stimuli and Paradigm
4.4. Data Analysis
4.4.1. ASSR Analysis
4.4.2. ERP Analysis
4.5. Molecular Genetic Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | Autism Spectrum Disorders |
| ASSR | Auditory Steady State Response |
| EEG | Electroencephalogram |
| ERP | Event-related potential |
| GABA | Gamma-aminobutyric acid |
| TD | Typically developing children |
| PMS | Phelan-McDermid Syndrome |
| SRS | Social Responsiveness Scale |
Appendix A
Individual ASSRs and ERPs


Appendix B
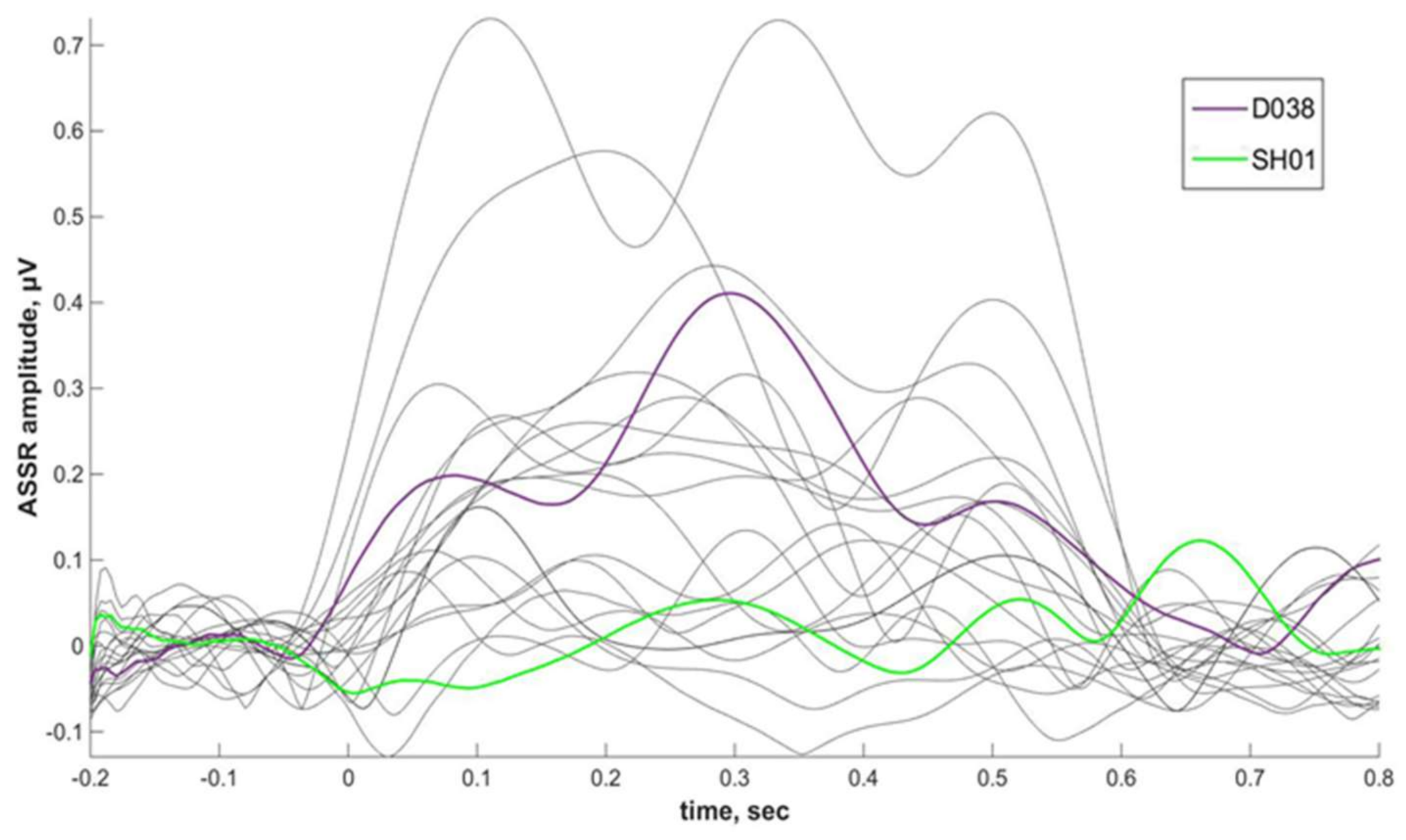
Phenibut effect on ASSR

Appendix C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ‘Old’ Group | ‘Young’ Group | ||||||
|---|---|---|---|---|---|---|---|
| Participant | Age | Sex | SRS, T-Scores | Participant | Age | Sex | SRS, T-Scores |
| D015 | 12.65 | f | 16 | D043 | 2.58 | f | 26 |
| D020 | 12.68 | f | 39 | D033 | 3.04 | m | |
| D021 | 13.45 | f | D007 | 5.04 | f | ||
| D041 | 13.94 | m | 56 | D009 | 5.86 | f | 22 |
| D002 | 14.10 | f | 42 | D022 | 6.01 | m | 23 |
| D025 | 14.47 | m | 24 | D012 | 6.42 | f | 21 |
| D027 | 14.76 | m | 22 | D004 | 7.93 | f | 15 |
| D047 | 15.18 | m | 35 | D001 | 8.18 | f | 28 |
| D046 | 15.4 | m | 52 | D006 | 8.24 | f | 41 |
| D048 | 15.74 | m | 49 | D013 | 8.4 | f | 22 |
| D049 | 17, 98 | f | 38 | D038 | 8.41 | m | 19 |
| D050 | 17.98 | f | 37 | D011 | 9.04 | f | 47 |
| D030 | 17.99 | f | D035 | 9.13 | m | 62 | |
| D016 | 9.43 | f | 11 | ||||
| D045 | 9.6 | m | 12 | ||||
| D017 | 9.83 | f | |||||
| D014 | 10.69 | f | |||||
| D003 | 11.98 | f | 37 | ||||
| D023 | 12.04 | f | 24 | ||||

References
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a Novel Family of Postsynaptic Density Proteins that Binds to the NMDA Receptor/PSD-95/GKAP Complex and Cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef]
- Sheng, M.; Kim, E. The Shank Family of Scaffold Proteins. J. Cell Sci. 2000, 113, 1851–1856. [Google Scholar] [PubMed]
- Phelan, K.; McDermid, H. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndr. 2011, 2, 186–201. [Google Scholar] [CrossRef]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Chen, C.-F.; Sharp, J.L.; Rollins, J.D.; Collins, J.S.; Rogers, R.C.; Phelan, K.; Dupont, B.R. 22q13.2q13.32 Genomic Regions Associated with Severity of Speech Delay, Developmental Delay, and Physical Features in Phelan–McDermid Syndrome. Genet. Med. 2013, 16, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.L.; Wong, A.C.C.; Shaw, S.R.; Tse, W.-Y.; Stapleton, A.G.; Phelan, M.C.; Hu, S.; Marshall, J.; McDermid, H.E. Molecular Characterisation of the 22q13 Deletion Syndrome Supports the Role of Haploinsufficiency of SHANK3/PROSAP2 in the Major Neurological Symptoms. J. Med. Genet. 2003, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Bassell, J.; Srivastava, S.; Prohl, A.K.; Scherrer, B.; Kapur, K.; Filip-Dhima, R.; Berry-Kravis, E.; Soorya, L.; Thurm, A.; Powell, C.M.; et al. Diffusion Tensor Imaging Abnormalities in the Uncinate Fasciculus and Inferior Longitudinal Fasciculus in Phelan-McDermid Syndrome. Pediatr. Neurol. 2020, 106, 24–31. [Google Scholar] [CrossRef]
- Costales, J.L.; Kolevzon, A. Phelan–McDermid Syndrome and SHANK3: Implications for Treatment. Neurotherapeutics 2015, 12, 620–630. [Google Scholar] [CrossRef]
- Harony-Nicolas, H.; De Rubeis, S.; Kolevzon, A.; Buxbaum, J.D. Phelan McDermid Syndrome. J. Child Neurol. 2015, 30, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Buxbaum, J.D. SHANK3 Haploinsufficiency: A “Common” but Underdiagnosed Highly Penetrant Monogenic Cause of Autism Spectrum Disorders. Mol. Autism 2013, 4, 17. [Google Scholar] [CrossRef]
- Oberman, L.M.; Rotenberg, A.; Pascual-Leone, A. Use of Transcranial Magnetic Stimulation in Autism Spectrum Disorders. J. Autism Dev. Disord. 2013, 45, 524–536. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-Analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, X.; Li, X.-L.; Towers, A.; Cao, X.; Wang, P.; Bowman, R.; Yang, H.; Goldstein, J.; Li, Y.-J.; et al. Epigenetic Dysregulation of SHANK3 in Brain Tissues from Individuals with Autism Spectrum Disorders. Hum. Mol. Genet. 2014, 23, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Schmeisser, M.J.; Schoen, M.; Boeckers, T.M. Postsynaptic ProSAP/Shank Scaffolds in the Cross-Hair of Synaptopathies. Trends Cell Biol. 2011, 21, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-Like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Zhao, Y.; Jaber, V.; Cong, L.; Lukiw, W.J. Deficits in the Proline-Rich Synapse-Associated Shank3 Protein in Multiple Neuropsychiatric Disorders. Front. Neurol. 2017, 8, 670. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Feng, P.M.G. SHANK Proteins: Roles at the Synapse and in Autism Spectrum Disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.-A.; Gao, X.; Fu, Z.; Feng, Y.M.P.M.Y.Z.J.-A.K.X.G.Z.F.G. Adult Restoration of Shank3 Expression Rescues Selective Autistic-like Phenotypes. Nat. Cell Biol. 2016, 530, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank Synaptic Scaffold Proteins: Keys to Understanding the Pathogenesis of Autism and Other Synaptic Disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Moessner, R.; Marshall, C.R.; Sutcliffe, J.S.; Skaug, J.; Pinto, D.; Vincent, J.; Zwaigenbaum, L.; Fernandez, B.; Roberts, W.; Szatmari, P.; et al. Contribution of SHANK3 Mutations to Autism Spectrum Disorder. Am. J. Hum. Genet. 2007, 81, 1289–1297. [Google Scholar] [CrossRef]
- Han, K.; Jr, J.L.H.; Schaaf, C.P.; Lu, H.-C.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 Overexpression Causes Manic-Like Behaviour with Unique Pharmacogenetic Properties. Nat. Cell Biol. 2013, 503, 72–77. [Google Scholar] [CrossRef]
- Failla, P.; Romano, C.; Alberti, A.; Vasta, A.; Buono, S.; Castiglia, L.; Luciano, D.; Di Benedetto, D.; Fichera, M.; Galesi, O. Schizophrenia in a Patient with Subtelomeric Duplication of Chromosome 22q. Clin. Genet. 2007, 71, 599–601. [Google Scholar] [CrossRef]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the Gene Encoding the Synaptic Scaffolding Protein shank3 Are Associated with Autism Spectrum Disorders. Nat. Genet. 2006, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, M.; Haugen, I.B.; Bakken, T.L.; Braaten, Ø. A 22q13.33 Duplication Harbouring the SHANK3 Gene: Does It Cause Neuropsychiatric Disorders? BMJ Case Rep. 2019, 12, e228258. [Google Scholar] [CrossRef] [PubMed]
- Ujfalusi, A.; Nagy, O.; Bessenyei, B.; Lente, G.; Kántor, I.; Borbély, Á.J.; Szakszon, K. 22q13 Microduplication Syndrome in Siblings with Mild Clinical Phenotype: Broadening the Clinical and Behavioral Spectrum. Mol. Syndr. 2020, 11, 146–152. [Google Scholar] [CrossRef]
- Kouser, M.; Speed, H.E.; Dewey, C.M.; Reimers, J.M.; Widman, A.J.; Gupta, N.; Liu, S.; Jaramillo, T.C.; Bangash, M.; Xiao, B.; et al. Loss of Predominant Shank3 Isoforms Results in Hippocampus-Dependent Impairments in Behavior and Synaptic Transmission. J. Neurosci. 2013, 33, 18448–18468. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chung, C.; Ha, S.; Lee, D.; Kim, D.-Y.; Kim, H.; Kim, E. Shank3-Mutant Mice Lacking Exon 9 Show Altered Excitation/Inhibition Balance, Enhanced Rearing, and Spatial Memory Deficit. Front. Cell. Neurosci. 2015, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Speed, H.E.; Kouser, M.; Xuan, Z.; Reimers, J.M.; Ochoa, C.F.; Gupta, N.; Liu, S.; Powell, C.M. Autism-Associated Insertion Mutation (InsG) of Shank3 Exon 21 Causes Impaired Synaptic Transmission and Behavioral Deficits. J. Neurosci. 2015, 35, 9648–9665. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.C.; Speed, H.E.; Xuan, Z.; Reimers, J.M.; Liu, S.; Powell, C.M. Altered Striatal Synaptic Function and Abnormal Behaviour inShank3Exon4-9 Deletion Mouse Model of Autism. Autism Res. 2016, 9, 350–375. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-H.; Ehlers, M.D. Modeling Autism by SHANK Gene Mutations in Mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Bakes, J.; Bradley, C.; Collingridge, G.L.; Kaang, B.-K. Shank Mutant Mice as an Animal Model of Autism. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130143. [Google Scholar] [CrossRef]
- Schmeisser, M.J. Translational Neurobiology in Shank Mutant Mice—Model Systems for Neuropsychiatric Disorders. Ann. Anat. Anat. Anz. 2015, 200, 115–117. [Google Scholar] [CrossRef]
- Dhamne, S.C.; Silverman, J.L.; Super, C.E.; Lammers, S.H.T.; Hameed, M.Q.; Modi, M.E.; Copping, N.A.; Pride, M.C.; Smith, D.G.; Rotenberg, A.; et al. Replicable in Vivo Physiological and Behavioral Phenotypes of the Shank3B Null Mutant Mouse Model of Autism. Mol. Autism 2017, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-Y.; Pang, K.; Kim, J.Y.; Ryu, J.R.; Kang, H.; Liu, Z.; Kim, W.-K.; Sun, W.; Kim, H.; Han, K. Post-transcriptional Regulation of SHANK3 Expression by MicroRNAs Related to Multiple Neuropsychiatric Disorders. Mol. Brain 2015, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, E.; Kim, R.; Kim, J.; Lee, S.; Park, H.; Yang, E.; Kim, H.; Kim, E. Shank2 Deletion in Parvalbumin Neurons Leads to Moderate Hyperactivity, Enhanced Self-Grooming and Suppressed Seizure Susceptibility in Mice. Front. Mol. Neurosci. 2018, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Filice, F.; Vörckel, K.J.; Sungur, A.Ö.; Wöhr, M.; Schwaller, B. Reduction in Parvalbumin Expression Not Loss of the Parvalbumin-Expressing GABA Interneuron Subpopulation in Genetic Parvalbumin and Shank Mouse Models of Autism. Mol. Brain 2016, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chen, Q.; Zhou, T.; Bozic, D.; Fu, Z.; Pan, J.Q.; Feng, G. Micro-Electrode Array Recordings Reveal Reductions in Both Excitation and Inhibition in Cultured Cortical Neuron Networks Lacking Shank3. Mol. Psychiatry 2015, 21, 159–168. [Google Scholar] [CrossRef]
- Bartos, M.; Vida, I.; Jonas, P. Synaptic Mechanisms of Synchronized Gamma Oscillations in Inhibitory Interneuron Networks. Nat. Rev. Neurosci. 2007, 8, 45–56. [Google Scholar] [CrossRef]
- Vinck, M.; Womelsdorf, T.; Buffalo, E.A.; DeSimone, R.; Fries, P. Attentional Modulation of Cell-Class-Specific Gamma-Band Synchronization in Awake Monkey Area V4. Neuron 2013, 80, 1077–1089. [Google Scholar] [CrossRef]
- Carlen, M.; Meletis, K.; Siegle, J.; Cardin, J.; Futai, K.; Vierling-Claassen, D.; Rühlmann, C.; Jones, S.R.; Deisseroth, K.; Sheng, M.; et al. A Critical Role for NMDA Receptors in Parvalbumin Interneurons for Gamma Rhythm Induction and Behavior. Mol. Psychiatry 2011, 17, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, N.; Schoffelen, J.-M.; Oostenveld, R.; Parkes, L.M.; Fries, P. Localizing Human Visual Gamma-Band Activity in Frequency, Time and Space. NeuroImage 2006, 29, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.; Picton, T.W.; Pantev, C. Temporal Integration in the Human Auditory Cortex as Represented by the Development of the Steady-State Magnetic Field. Hear. Res. 2002, 165, 68–84. [Google Scholar] [CrossRef]
- Ross, B.; Pantev, C. Auditory Steady-State Responses Reveal Amplitude Modulation Gap Detection Thresholds. J. Acoust. Soc. Am. 2004, 115, 2193–2206. [Google Scholar] [CrossRef] [PubMed]
- Onitsuka, T.; Oribe, N.; Nakamura, I.; Kanba, S. Review of Neurophysiological Findings in Patients with Schizophrenia. Psychiatry Clin. Neurosci. 2013, 67, 461–470. [Google Scholar] [CrossRef]
- Picton, T.W.; John, M.S.; Dimitrijevic, A.; Purcell, D. Human Auditory Steady-State Responses: Respuestas Auditivas de Estado Estable en Humanos. Int. J. Audiol. 2003, 42, 177–219. [Google Scholar] [CrossRef] [PubMed]
- Tateno, T.; Harsch, A.; Robinson, H.P.C. Threshold Firing Frequency–Current Relationships of Neurons in Rat Somatosensory Cortex: Type 1 and Type 2 Dynamics. J. Neurophysiol. 2004, 92, 2283–2294. [Google Scholar] [CrossRef]
- Golomb, D.; Donner, K.; Shacham, L.; Shlosberg, D.; Amitai, Y.; Hansel, D. Mechanisms of Firing Patterns in Fast-Spiking Cortical Interneurons. PLoS Comput. Biol. 2007, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Enakao, K.; Enakazawa, K. Brain State-Dependent Abnormal LFP Activity in the Auditory Cortex of a Schizophrenia Mouse Model. Front. Neurosci. 2014, 8, 168. [Google Scholar] [CrossRef]
- Sivarao, D.V.; Chen, P.; Senapati, A.; Yang, Y.; Fernandes, A.; Benitex, Y.; Whiterock, V.; Li, Y.-W.; Ahlijanian, M.K. 40 Hz Auditory Steady-State Response Is a Pharmacodynamic Biomarker for Cortical NMDA Receptors. Neuropsychopharmacology 2016, 41, 2232–2240. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.M.; Timi, M.P.; Hong, L.E.; O’Donnell, P. Effects of NMDA and GABA-A Receptor Antagonism on Auditory Steady-State Synchronization in Awake Behaving Rats. Int. J. Neuropsychopharmacol. 2015, 18, pyu118. [Google Scholar] [CrossRef] [PubMed]
- Leishman, E.; O’Donnell, B.F.; Millward, J.B.; Vohs, J.L.; Rass, O.; Krishnan, G.P.; Bolbecker, A.R.; Morzorati, S.L. Phencyclidine Disrupts the Auditory Steady State Response in Rats. PLoS ONE 2015, 10, e0134979. [Google Scholar] [CrossRef]
- Sivarao, D.V.; Frenkel, M.; Chen, P.; Healy, F.L.; Lodge, N.J.; Zaczek, R. MK-801 Disrupts and Nicotine Augments 40 Hz Auditory Steady State Responses in the Auditory Cortex of the Urethane-Anesthetized Rat. Neuropharmacology 2013, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kirli, K.K.; Ermentrout, G.B.; Cho, R.Y.; Kirli, K.K. Computational Study of NMDA Conductance and Cortical Oscillations in Schizophrenia. Front. Comput. Neurosci. 2014, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.M. The Functional Consequences of Cortical Circuit Abnormalities on Gamma Oscillations in Schizophrenia: Insights from Computational Modeling. Front. Hum. Neurosci. 2009, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Thuné, H.; Recasens, M.; Uhlhaas, P.J. The 40-Hz Auditory Steady-State Response in Patients with Schizophrenia. JAMA Psychiatry 2016, 73, 1145–1153. [Google Scholar] [CrossRef]
- Parker, D.A.; Hamm, J.P.; McDowell, J.E.; Keedy, S.K.; Gershon, E.S.; Ivleva, E.I.; Pearlson, G.D.; Keshavan, M.S.; Tamminga, C.A.; Sweeney, J.A.; et al. Auditory Steady-State EEG Response Across the Schizo-Bipolar Spectrum. Schizophr. Res. 2019, 209, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Isomura, S.; Onitsuka, T.; Tsuchimoto, R.; Nakamura, I.; Hirano, S.; Oda, Y.; Oribe, N.; Hirano, Y.; Ueno, T.; Kanba, S. Differentiation between Major Depressive Disorder and Bipolar Disorder by Auditory Steady-State Responses. J. Affect. Disord. 2016, 190, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Rass, O.; Krishnan, G.; Brenner, A.C.; Hetrick, W.P.; Merrill, C.C.; Shekhar, A.; O’Donnell, B.F. Auditory Steady State Response in Bipolar Disorder: Relation to Clinical State, Cognitive Performance, Medication Status, and Substance Disorders. Bipolar Disord. 2010, 12, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Onitsuka, T.; Tsuchimoto, R.; Hirano, S.; Oribe, N.; Ueno, T.; Hirano, Y.; Nakamura, I.; Miura, T.; Kanba, S. Gamma Band Neural Synchronization Deficits for Auditory Steady State Responses in Bipolar Disorder Patients. PLoS ONE 2012, 7, e39955. [Google Scholar] [CrossRef]
- Rojas, D.C.; Teale, P.D.; Maharajh, K.; Kronberg, E.; Youngpeter, K.; Wilson, L.B.; Wallace, A.; Hepburn, S. Transient and Steady-State Auditory Gamma-Band Responses in First-Degree Relatives of People with Autism Spectrum Disorder. Mol. Autism 2011, 2, 11. [Google Scholar] [CrossRef]
- Seymour, R.A.; Rippon, G.; Gooding-Williams, G.; Sowman, P.F.; Kessler, K. Reduced Auditory Steady State Responses in Autism Spectrum Disorder. Mol. Autism 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Hong, L.E. Evoked Gamma Band Synchronization and the Liability for Schizophrenia*1. Schizophr. Res. 2004, 70, 293–302. [Google Scholar] [CrossRef]
- Ono, Y.; Kudoh, K.; Ikeda, T.; Takahashi, T.; Yoshimura, Y.; Minabe, Y.; Kikuchi, M. Auditory Steady-State Response at 20 Hz and 40 Hz in Young Typically Developing Children and Children with Autism Spectrum Disorder. Psychiatry Clin. Neurosci. 2020, 74, 354–361. [Google Scholar] [CrossRef]
- Stroganova, T.A.; Komarov, K.S.; Sysoeva, O.V.; Goiaeva, D.E.; Obukhova, T.S.; Ovsiannikova, T.M.; Prokofyev, A.O.; Orekhova, E.V. Left Hemispheric Deficit in the Sustained Neuromagnetic Response to Periodic Click Trains in Children with ASD. Mol. Autism 2020, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Haenschel, C.; Nikolić, D.; Singer, W. The Role of Oscillations and Synchrony in Cortical Networks and Their Putative Relevance for the Pathophysiology of Schizophrenia. Schizophr. Bull. 2008, 34, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.; Fujioka, T. 40-Hz Oscillations Underlying Perceptual Binding in Young and Older Adults. Psychophysiology 2016, 53, 974–990. [Google Scholar] [CrossRef] [PubMed]
- Baltus, A.; Herrmann, C.S. Auditory Temporal Resolution Is Linked to Resonance Frequency of the Auditory Cortex. Int. J. Psychophysiol. 2015, 98, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Light, G.A.; Hsu, J.L.; Hsieh, M.H.; Meyer-Gomes, K.; Sprock, J.; Swerdlow, N.R.; Braff, D.L. Gamma Band Oscillations Reveal Neural Network Cortical Coherence Dysfunction in Schizophrenia Patients. Biol. Psychiatry 2006, 60, 1231–1240. [Google Scholar] [CrossRef]
- Tada, M.; Nagai, T.; Kirihara, K.; Koike, S.; Suga, M.; Araki, T.; Kobayashi, T.; Kasai, K. Differential Alterations of Auditory Gamma Oscillatory Responses Between Pre-Onset High-Risk Individuals and First-Episode Schizophrenia. Cereb. Cortex 2016, 26, 1027–1035. [Google Scholar] [CrossRef]
- Koshiyama, D.; Kirihara, K.; Tada, M.; Nagai, T.; Fujioka, M.; Ichikawa, E.; Ohta, K.; Tani, M.; Tsuchiya, M.; Kanehara, A.; et al. Electrophysiological Evidence for Abnormal Glutamate-GABA Association Following Psychosis Onset. Transl. Psychiatry 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Filimonenko, Y.I.; Timofeev, V.I. Wechsler Intelligence Scale for Children. Methodological Manual; Imaton: Saint Petersburg, Russia, 2006; p. 112. (in Russian) [Google Scholar]
- Constantino, J.N.; Davis, S.A.; Todd, R.D.; Schindler, M.K.; Gross, M.M.; Brophy, S.L.; Metzger, L.M.; Shoushtari, C.S.; Splinter, R.; Reich, W. Validation of a Brief Quantitative Measure of Autistic Traits: Comparison of the Social Responsiveness Scale with the Autism Diagnostic Interview-Revised. J. Autism Dev. Disord. 2003, 33, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism Diagnostic Interview-Revised: A Revised Version of a Diagnostic Interview for Caregivers of Individuals with Possible Pervasive Developmental Disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef]
- Philippe, A.; Boddaert, N.; Vaivre-Douret, L.; Robel, L.; Danon-Boileau, L.; Malan, V.; De Blois, M.-C.; Heron, D.; Colleaux, L.; Golse, B.; et al. Neurobehavioral Profile and Brain Imaging Study of the 22q13.3 Deletion Syndrome in Childhood. Pediatrics 2008, 122, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective Investigation of Autism and Genotype-Phenotype Correlations in 22q13 Deletion Syndrome and SHANK3 Deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Zwanenburg, R.J.; Ruiter, S.A.; Heuvel, E.R.V.D.; Flapper, B.C.; Van Ravenswaaij-Arts, C.M. Developmental Phenotype in Phelan-McDermid (22q13.3 Deletion) Syndrome: A Systematic and Prospective Study in 34 Children. J. Neurodev. Disord. 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Chen, H.-I.; Liao, H.-M.; Chen, Y.-J.; Fang, J.-S.; Lee, K.-F.; Gau, S.S.-F. Clinical and Molecular Characterization of Three Genomic Rearrangements at Chromosome 22q13.3 Associated with Autism Spectrum Disorder. Psychiatr. Genet. 2017, 27, 23–33. [Google Scholar] [CrossRef]
- Okamoto, N.; Kubota, T.; Nakamura, Y.; Murakami, R.; Nishikubo, T.; Tanaka, I.; Takahashi, Y.; Hayashi, S.; Imoto, I.; Inazawa, J.; et al. 22q13 Microduplication in Two Patients with Common Clinical Manifestations: A Recognizable Syndrome? Am. J. Med Genet. Part A 2007, 143, 2804–2809. [Google Scholar] [CrossRef]
- Destrée, A.; Hilbert, P.; Boulanger, S. A 273-kb Duplication at 22q13.33 Encompassing the Shank3 Gene in 2 Sibs with Microcephaly, Behavioral Disorder and Learning Disabilities. In Proceedings of European Society of Human Genetics Conference, Amsterdam, The Netherlands, 28–31 May 2011, P02.034. Eur. J. Hum. Genet. 2011, 19, 76. [Google Scholar]
- Sarasua, S.M.; Boccuto, L.; Sharp, J.L.; Dwivedi, A.; Chen, C.-F.; Rollins, J.D.; Rogers, R.C.; Phelan, K.; Dupont, B.R. Clinical and Genomic Evaluation of 201 Patients with Phelan–McDermid Syndrome. Qual. Life Res. 2014, 133, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Kolevzon, A.; Angarita, B.; Bush, L.; Wang, A.T.; Frank, Y.; Yang, A.; Rapaport, R.; Saland, J.; Srivastava, S.; Farrell, C.; et al. Phelan-McDermid Syndrome: A Review of the Literature and Practice Parameters for Medical Assessment and Monitoring. J. Neurodev. Disord. 2014, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Reierson, G.; Bernstein, J.; Froehlich-Santino, W.; Urban, A.; Purmann, C.; Berquist, S.; Jordan, J.; O’Hara, R.; Hallmayer, J. Characterizing Regression in Phelan McDermid Syndrome (22q13 Deletion Syndrome). J. Psychiatr. Res. 2017, 91, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Kolevzon, A.; Delaby, E.; Berry-Kravis, E.; Buxbaum, J.D.; Betancur, C. Neuropsychiatric Decompensation in Adolescents and Adults with Phelan-McDermid Syndrome: A Systematic Review of the Literature. Mol. Autism 2019, 10, 1–22. [Google Scholar] [CrossRef]
- Hileman, C.M.; Henderson, H.A.; Mundy, P.; Newell, L.C.; Jaime, M. Developmental and Individual Differences on the P1 and N170 ERP Components in Children with and without Autism. Dev. Neuropsychol. 2011, 36, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Itier, R.J.; Taylor, M.J. Effects of Repetition and Configural Changes on the Development of Face Recognition Processes. Dev. Sci. 2004, 7, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Reese, M. Effects of Age, Gender, and Genotype on Auditory Processing in Phelan-McDermid Syndrome. Master’s Thesis, University of Oklahoma, Norman, OK, USA, 2019. [Google Scholar]
- Brittenham, C. Objective Measures of Electrophysiological Responses of Children with Idiopathic Autism Spectrum Disorder and Phelan-McDermid Syndrome to a Contrast-Reversing Checkerboard. Master’s Thesis, Hunter College, University of New York, New York, NY, USA, 18 December 2017. [Google Scholar]
- Siper, P.; George-Jones, J.; Lurie, S.; Rowe, M.; Durkin, A.; Weissman, J.; Meyering, K.; Rouhandeh, A.; Buxbaum, J.; Kolevzon, A. 23. Biomarker Discovery in ASD: Visual Evoked Potentials as a Biomarker of Phelan-McDermid Syndrome. Biol. Psychiatry 2018, 83, S9. [Google Scholar] [CrossRef]
- Whitehouse, A.J.; Bishop, D.V. Do Children with Autism ‘Switch off’ to Speech Sounds? An Investigation Using Event-Related Potentials. Dev. Sci. 2008, 11, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Stroganova, T.A.; Kozunov, V.V.; Posikera, I.N.; Galuta, I.A.; Gratchev, V.V.; Orekhova, E.V. Abnormal Pre-Attentive Arousal in Young Children with Autism Spectrum Disorder Contributes to Their Atypical Auditory Behavior: An ERP Study. PLoS ONE 2013, 8, e69100. [Google Scholar] [CrossRef] [PubMed]
- Madsen, G.F.; Bilenberg, N.; Jepsen, J.R.; Glenthøj, B.; Cantio, C.; Oranje, B. Normal P50 Gating in Children with Autism, Yet Attenuated P50 Amplitude in the Asperger Subcategory. Autism Res. 2015, 8, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Isenstein, E.; Durkin, A.; Zhang, Y.; Feldman, E.; Servedio, N.; Harony-Nicolas, H.; Buxbaum, J.; Kolevzon, A.; Siper, P.; Foss-Feig, J. T185. Electrophysiological Evidence of Auditory Habituation Abnormalities in Young Adults with Phelan-McDermid Syndrome. Biol. Psychiatry 2018, 83, S200. [Google Scholar] [CrossRef]
- Ponson, L.; Gomot, M.; Blanc, R.; Barthelemy, C.; Roux, S.; Munnich, A.; Romana, S.; Aguillon-Hernandez, N.; Malan, V.; Bonnet-Brilhault, F. 22q13 Deletion Syndrome: Communication Disorder or Autism? Evidence from a Specific Clinical and Neurophysiological Phenotype. Transl. Psychiatry 2018, 8, 146. [Google Scholar] [CrossRef]
- Griskova-Bulanova, I.; Dapsys, K.; Maciulis, V. Does Brain Ability to Synchronize with 40 Hz Auditory Stimulation Change with Age? Acta Neurobiol. Exp. 2013, 73, 564–570. [Google Scholar]
- Poulsen, C.; Picton, T.W.; Paus, T. Age-Related Changes in Transient and Oscillatory Brain Responses to Auditory Stimulation during Early Adolescence. Dev. Sci. 2009, 12, 220–235. [Google Scholar] [CrossRef]
- Maurizi, M.; Almadori, G.; Paludetti, G.; Ottaviani, F.; Rosignoli, M.; Lucianob, R. 40-Hz Steady-State Responses in Newborns and in Children. Int. J. Audiol. 1990, 29, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Stapells, D.; Herdman, A.; Small, S.; Dimitrijevic, A.; Hatton, J. Current Status of the Auditory Steady-State Responses for Estimating an Infant’s Audiogram; Phonak: Zurich, Sweden, 2005; pp. 43–59. [Google Scholar]
- Engineer, C.T.; Rahebi, K.C.; Borland, M.S.; Buell, E.P.; Im, K.W.; Wilson, L.G.; Sharma, P.; Vanneste, S.; Harony-Nicolas, H.; Buxbaum, J.D.; et al. Shank3-Deficient Rats Exhibit Degraded Cortical Responses to Sound. Autism Res. 2017, 11, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Griškova-Bulanova, I.; Rukšėnas, O.; Dapšys, K.; Mačiulis, V.; Arnfred, S.M. Distraction Task Rather Than Focal Attention Modulates Gamma Activity Associated with Auditory Steady-State Responses (ASSRs). Clin. Neurophysiol. 2011, 122, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Griskova, I.; Morup, M.; Parnas, J.; Ruksenas, O.; Arnfred, S.M. The Amplitude and Phase Precision of 40 Hz Auditory Steady-State Response Depend on the Level of Arousal. Exp. Brain Res. 2007, 183, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bensmann, W.; Vahid, A.; Beste, C.; Stock, A.-K. The Intensity of Early Attentional Processing, but Not Conflict Monitoring, Determines the Size of Subliminal Response Conflicts. Front. Hum. Neurosci. 2019, 13, 13. [Google Scholar] [CrossRef]
- Martin, B.A.; Tremblay, K.L.; Korczak, P. Speech Evoked Potentials: From the Laboratory to the Clinic. Ear Hear. 2008, 29, 285–313. [Google Scholar] [CrossRef]
- Oostenveld, R.; Fries, P.; Maris, E.; Schoffelen, J.-M. FieldTrip: Open Source Software for Advanced Analysis of MEG, EEG, and Invasive Electrophysiological Data. Comput. Intell. Neurosci. 2010, 2011, 1–9. [Google Scholar] [CrossRef]
- Griskova, I.; Morup, M.; Parnas, J.; Rukšėnas, O.; Arnfred, S.M.; Griškova-Bulanova, I. Two Discrete Components of the 20 Hz Steady-State Response Are Distinguished through the Modulation of Activation Level. Clin. Neurophysiol. 2009, 120, 904–909. [Google Scholar] [CrossRef]
- Hirano, Y.; Nakamura, I.; Tamura, S.; Onitsuka, T. Long-Term Test-Retest Reliability of Auditory Gamma Oscillations Between Different Clinical EEG Systems. Front. Psychiatry 2020, 11, 11. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Korostelev, S.A.; Zelenova, M.A.; Yurov, Y.B. Long Contiguous Stretches of Homozygosity Spanning Shortly the Imprinted Loci Are Associated with Intellectual Disability, Autism and/or Epilepsy. Mol. Cytogenet. 2015, 8, 77. [Google Scholar] [CrossRef][Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Zelenova, M.A.; Kurinnaia, O.S.; Vasin, K.S.; Kutsev, S.I. The Cytogenomic “Theory of Everything”: Chromohelkosis May Underlie Chromosomal Instability and Mosaicism in Disease and Aging. Int. J. Mol. Sci. 2020, 21, 8328. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Voinova, V.Y.; Yurov, Y.B. 3p22.1p21.31 Microdeletion Identifies CCK as Asperger Syndrome Candidate Gene and Shows the Way for Therapeutic Strategies in Chromosome Imbalances. Mol. Cytogenet. 2015, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. In Silico Molecular Cytogenetics: A Bioinformatic Approach to Prioritization of Candidate Genes and Copy Number Variations for Basic and Clinical Genome Research. Mol. Cytogenet. 2014, 7, 98. [Google Scholar] [CrossRef] [PubMed]







| ASSR, μV | P1, μV | N1, μV | P2, μV | |
|---|---|---|---|---|
| SH01 (15.1 y o) | −0.002 | 1.359 | 1.102 | 3.670 |
| Old group (16.1 ± 1.9 y o) | 0.274 ± 0.103 | 3.159 ± 2.017 | 1.829 ± 1.885 | 2.332 ± 2.241 |
| Young group (7.8 ± 2.6 y o) | 0.158 ± 0.155 | 4.267 ± 1.886 | 6.059 ± 3.801 | 6.455 ± 4.754 |
| Language Problem | Mental Retardation | Autism | Microcephaly | Seizures | Attention Deficit | Affective/ Psychiatric Symptoms | Physical Abnormalities/ Dysmorphism | |
|---|---|---|---|---|---|---|---|---|
| SH01, our patient | mild dyslexia, dysgraphia | mild, IQ 64 | SRS = 63, no diagnosis | + | − | + | irritability, aggressiveness | elongated skull, protruding auricles, sandal gap |
| Johannessen et al. (2019) [23] | n/a | mild | n/a | n/a | + | + | bipolar disease | Tourette syndrome (motor tics), mild dysmorphism |
| Han et al. (2013) [20] Patient 1 | n/a | mild | n/a | n/a | + | + | destructive behavior | dysmorphism |
| Han et al. (2013) [20] Patient 2 | n/a | mild | n/a | n/a | + | + | bipolar disorder | − |
| Chen et al. (2017) [76] | language delay, echolalia | mild, IQ 62 | autistic traits, no diagnosis | n/a | n/a | + | irritability | − |
| Ujfalusi et al. (2020) [24] Patient 1 | − | mild, IQ 72 | n/a | − | − | n/a | − | dysmorphism |
| Ujfalusi et al. (2020) [24] Patient 2 | dyslexia, dysgraphia | mild, IQ 79 | n/a | − | + | − | bipolar disorder, temper tantrums | dysmorphism |
| Okamoto et al. (2007) [77] Patient 1 | language delay | moderate, DQ 40 | − | − | - | − | − | hypotonia, dysmorphism |
| Okamoto et al. (2007) [77] Patient 2 | language delay | moderate, DQ 46 | − | − | − | − | − | hypotonia, dysmorphism |
| Failla et al. (2007) [21] | incoherent speech | mild, IQ 73 | − | + | − | + | schizophrenia, irritability, aggressiveness | dysmorphism |
| Destrée et al. (2011) [78] Patient 1 | language delay | mild | n/a | + | n/a | n/a | n/a | dysmorphism, growth retardation |
| Destrée et al. (2011) [78] Patient 2 | language delay | mild | n/a | + | n/a | n/a | n/a | dysmorphism, growth retardation |
| Duplication (n = 31), % | 35% | 80% (from mild to severe) | 19% | 17% | 17% | 16% | bipolar disorder –4% psychosis−7% | Dysmorphism-54% |
| Deletion/PMS, % | 100% (no speech in 50%) [79] | 96% (with profound in 53%) [74] | 31–84% [74,79] | 6–14% [80] | 63% [81] | 11% [74] | bipolar disorder –54% psychosis–12% irritability–36% [82] | 68–93% dysmorphism [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neklyudova, A.K.; Portnova, G.V.; Rebreikina, A.B.; Voinova, V.Y.; Vorsanova, S.G.; Iourov, I.Y.; Sysoeva, O.V. 40-Hz Auditory Steady-State Response (ASSR) as a Biomarker of Genetic Defects in the SHANK3 Gene: A Case Report of 15-Year-Old Girl with a Rare Partial SHANK3 Duplication. Int. J. Mol. Sci. 2021, 22, 1898. https://doi.org/10.3390/ijms22041898
Neklyudova AK, Portnova GV, Rebreikina AB, Voinova VY, Vorsanova SG, Iourov IY, Sysoeva OV. 40-Hz Auditory Steady-State Response (ASSR) as a Biomarker of Genetic Defects in the SHANK3 Gene: A Case Report of 15-Year-Old Girl with a Rare Partial SHANK3 Duplication. International Journal of Molecular Sciences. 2021; 22(4):1898. https://doi.org/10.3390/ijms22041898
Chicago/Turabian StyleNeklyudova, Anastasia K., Galina V. Portnova, Anna B. Rebreikina, Victoria Yu Voinova, Svetlana G. Vorsanova, Ivan Y. Iourov, and Olga V. Sysoeva. 2021. "40-Hz Auditory Steady-State Response (ASSR) as a Biomarker of Genetic Defects in the SHANK3 Gene: A Case Report of 15-Year-Old Girl with a Rare Partial SHANK3 Duplication" International Journal of Molecular Sciences 22, no. 4: 1898. https://doi.org/10.3390/ijms22041898
APA StyleNeklyudova, A. K., Portnova, G. V., Rebreikina, A. B., Voinova, V. Y., Vorsanova, S. G., Iourov, I. Y., & Sysoeva, O. V. (2021). 40-Hz Auditory Steady-State Response (ASSR) as a Biomarker of Genetic Defects in the SHANK3 Gene: A Case Report of 15-Year-Old Girl with a Rare Partial SHANK3 Duplication. International Journal of Molecular Sciences, 22(4), 1898. https://doi.org/10.3390/ijms22041898