
Microtubule Destabilizing Sulfonamides as an Alternative to Taxane-Based Chemotherapy


 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Synthesis of MDS
2.2. Replacement of CA-4 Olefin by a Sulfonamide Highly Increased Aqueous Solubility
2.3. MDS Inhibit Cell Proliferation in Breast, Cervix, and Ovarian Tumor Cells
2.4. Lead Compounds Overcome MDR-Mediated Resistance
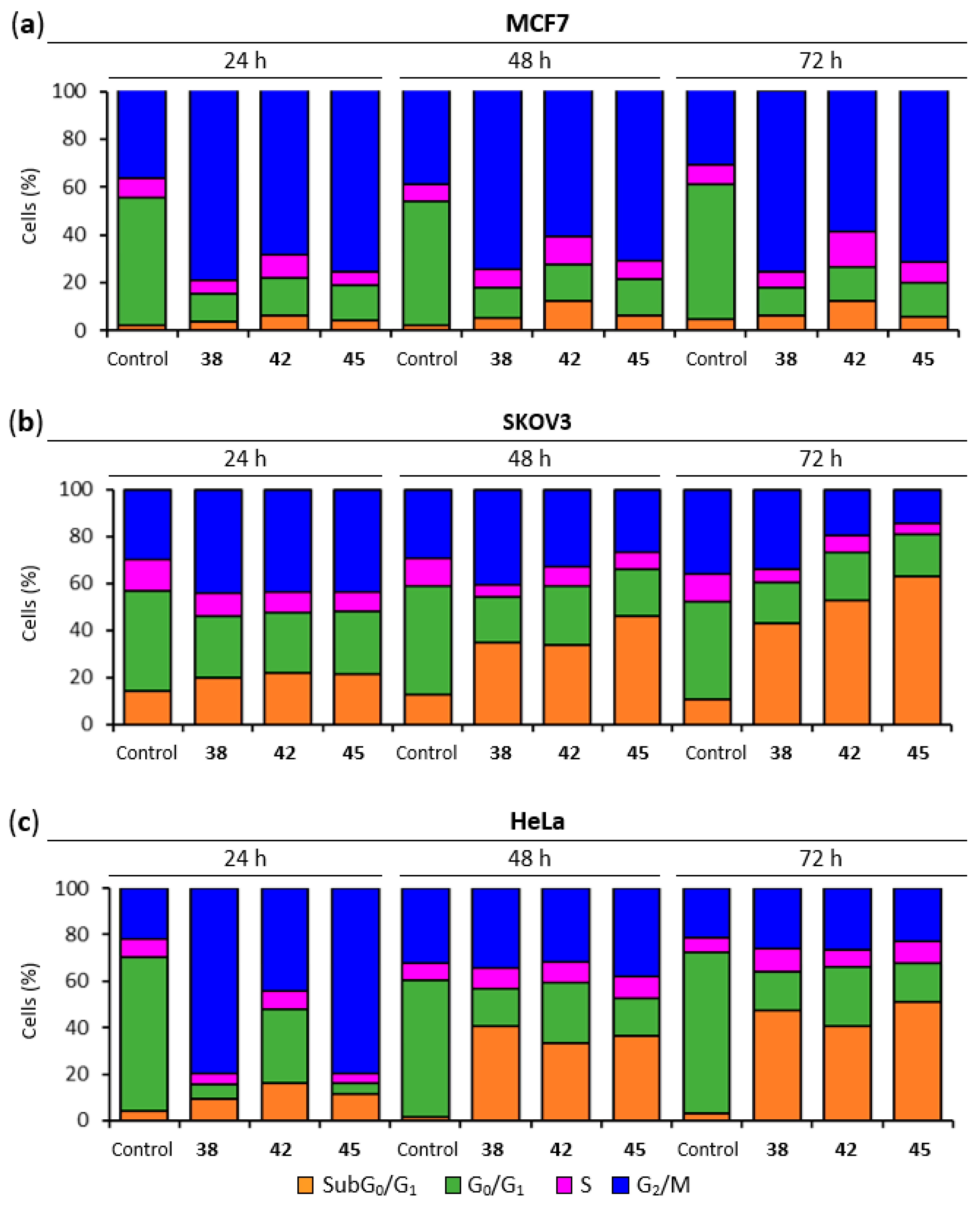
2.5. Lead Compounds Induce G2/M Arrest in Breast, Ovarian and Cervix Tumor Cells
2.6. Lead Compounds Trigger Apoptotic Cell Death
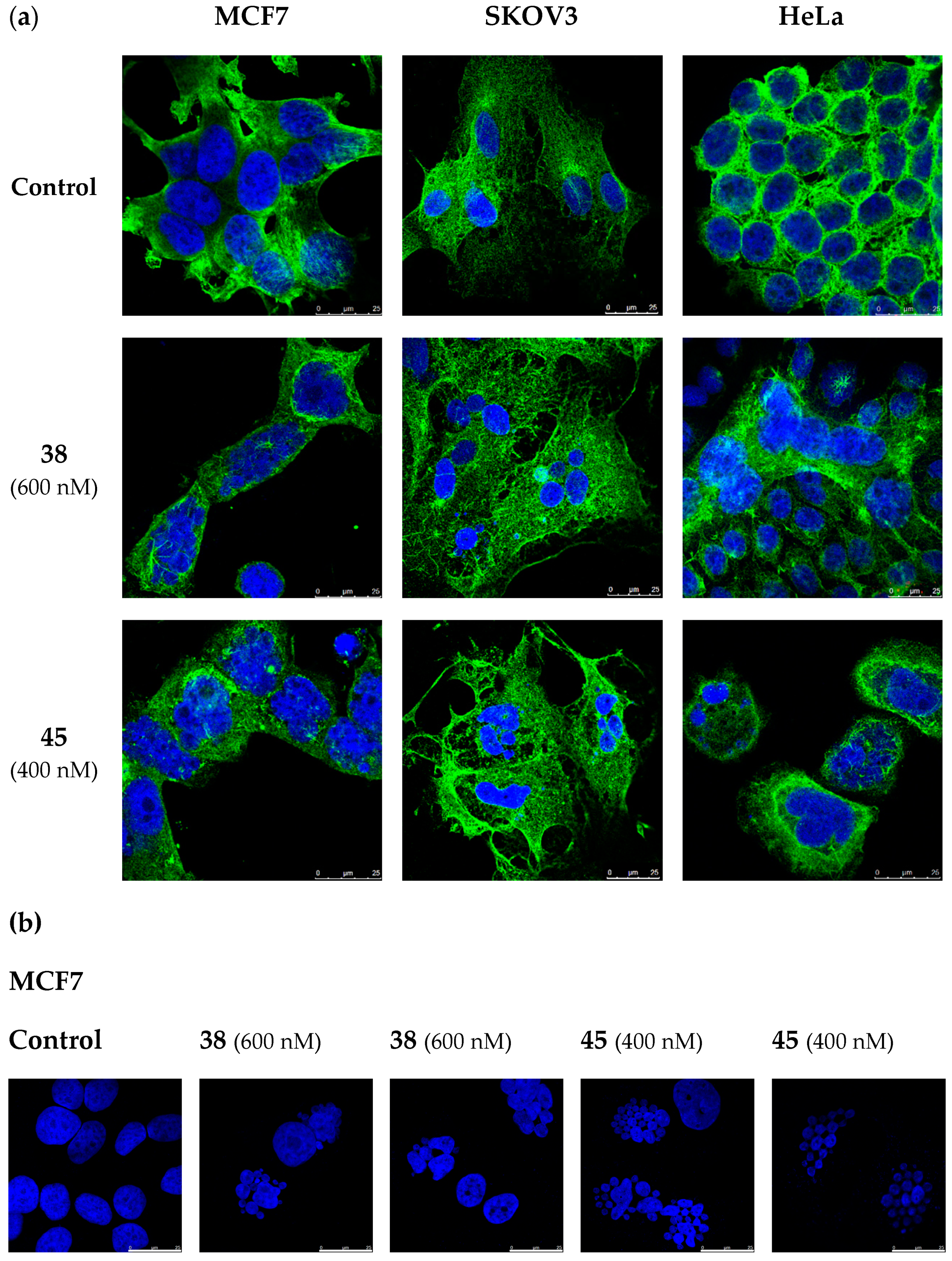
2.7. Lead Compounds Disrupt the Microtubule Network and Inhibit Tubulin Polymerization
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Chemical Synthesis
4.1.2. Chemical Characterization of Lead MDS 38, 42, and 45
4.1.3. Aqueous Solubility
4.2. Biological Evaluation
4.2.1. Cell Lines and Cell Culture Conditions
4.2.2. Cell Proliferation Assay
4.2.3. Cell Cycle Analysis
4.2.4. Apoptotic Cell Death Quantification
4.2.5. Immunofluorescence
4.2.6. Tubulin Isolation
4.2.7. Tubulin Polymerization Inhibition (TPI) Assay
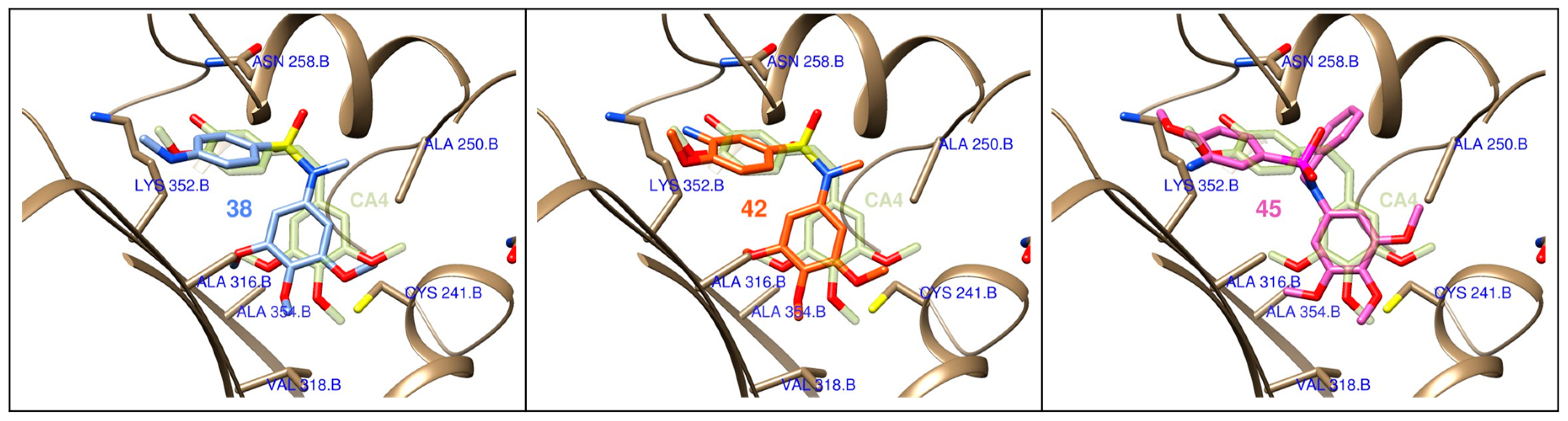
4.3. Computational Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.; Gomez, H. Chemotherapy resistance in metastatic breast cancer: The evolving role of ixabepilone. Breast Cancer Res. 2010, 12, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of Resistance to Systemic Therapy in Patients with Breast Cancer. In Breast Cancer Chemosensitivity; Madame Curie Biosci. Database; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299X1986081. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, P.; Asselin, E. Resistance to chemotherapy and hormone therapy in endometrial cancer. Endocr. Relat. Cancer 2009, 16, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Moxley, K.M.; McMeekin, D.S. Endometrial Carcinoma: A Review of Chemotherapy, Drug Resistance, and the Search for New Agents. Oncologist 2010, 15, 1026–1033. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/ (accessed on 27 May 2020).
- Ofir, R.; Seidman, R.; Rabinski, T.; Krup, M.; Yavelsky, V.; Weinstein, Y.; Wolfson, M. Taxol-induced apoptosis in human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase-3 and caspase-9 independent. Cell Death Differ. 2002, 9, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- McGuire, W.P.; Markman, M. Primary ovarian cancer chemotherapy: Current standards of care. Br. J. Cancer 2003, 89, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Bicaku, E.; Xiong, Y.; Marchion, D.C.; Chon, H.S.; Stickles, X.B.; Chen, N.; Judson, P.L.; Hakam, A.; Gonzalez-Bosquet, J.; Wenham, R.M.; et al. In vitro analysis of ovarian cancer response to cisplatin, carboplatin, and paclitaxel identifies common pathways that are also associated with overall patient survival. Br. J. Cancer 2012, 106, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Wilcken, N.; Simes, R.J. A systematic review of taxane-containing regimens for metastatic breast cancer. Br. J. Cancer 2005, 93, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrichs, K.; Hölzel, F.; Jänicke, F. Combination of taxanes and anthracyclines in first-line chemotherapy of metastatic breast cancer: An interim report. Eur. J. Cancer 2002, 38, 1730–1738. [Google Scholar] [CrossRef]
- Telli, M.L.; Carlson, R.W. First-line chemotherapy for metastatic breast cancer. Clin. Breast Cancer 2009, 9, S66–S72. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Burzykowski, T.; Buyse, M.; Sledge, G.; Carmichael, J.; Lück, H.J.; Mackey, J.R.; Nabholtz, J.M.; Paridaens, R.; Biganzoli, L.; et al. Taxanes alone or in combination with anthracyclines as first-line therapy of patients with metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, D.; Pectasides, E.; Economopoulos, T. Systemic therapy in metastatic or recurrent endometrial cancer. Cancer Treat. Rev. 2007, 33, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Serkies, K.; Jassem, J. Systemic therapy for cervical carcinoma—Current status. Chin. J. Cancer Res. 2018, 30, 209–221. [Google Scholar] [CrossRef]
- Boussios, S.; Seraj, E.; Zarkavelis, G.; Petrakis, D.; Kollas, A.; Kafantari, A.; Assi, A.; Tatsi, K.; Pavlidis, N.; Pentheroudakis, G. Management of patients with recurrent/advanced cervical cancer beyond first line platinum regimens: Where do we stand? A literature review. Crit. Rev. Oncol. Hematol. 2016, 108, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Watanabe, J.; Ishihara, K. Enhanced solubility of paclitaxel using water-soluble and biocompatible 2-methacryloyloxyethyl phosphorylcholine polymers. J. Biomed. Mater. Res. Part A 2003, 65, 209–214. [Google Scholar] [CrossRef]
- Hamada, H.; Ishihara, K.; Masuoka, N.; Mikuni, K.; Nakajima, N. Enhancement of water-solubility and bioactivity of paclitaxel using modified cyclodextrins. J. Biosci. Bioeng. 2006, 102, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Cui, F.-D.; Choi, M.-K.; Lin, H.; Chung, S.-J.; Shim, C.-K.; Kim, D.-D. Liposome Formulation of Paclitaxel with Enhanced Solubility and Stability. Drug Deliv. 2007, 14, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Eric, K.; Rowinsky, M.D.; Ross, C.; Donehower, M.D. Paclitaxel (Taxol®). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef]
- Hennenfent, K.L.; Govindan, R. Review Novel formulations of taxanes: A review. Old wine in a new bottle? Ann. Oncol. 2006, 17, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Ten Tije, A.J.; Verweij, J.; Loos, W.J.; Sparreboom, A. Pharmacological effects of formulation vehicles: Implications for cancer chemotherapy. Clin. Pharmacokinet. 2003, 42, 665–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Gligorov, J.; Lotz, J.P. Preclinical Pharmacology of the Taxanes: Implications of the Differences. Oncologist 2004, 9, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [Green Version]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, R.; Duan, Z.; Lamendola, D.; Penson, R.; Seiden, M. Paclitaxel Resistance: Molecular Mechanisms and Pharmacologic Manipulation. Curr. Cancer Drug Targets 2003, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.; Briasoulis, E.; Linardou, H.; Bafaloukos, D.; Papadimitriou, C. Taxane resistance in breast cancer: Mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 2012, 38, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, S.; Mangia, A.; Lacalamita, R.; Bellizzi, A.; Fedele, V.; Chiriatti, A.; Thomssen, C.; Kendzierski, N.; Latorre, A.; Lorusso, V.; et al. Cytoskeleton and paclitaxel sensitivity in breast cancer: The role of β-tubulins. Int. J. Cancer 2007, 120, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Chong, T.; Sarac, A.; Yao, C.Q.; Liao, L.; Lyttle, N.; Boutros, P.C.; Bartlett, J.M.S.; Spears, M. Deregulation of the spindle assembly checkpoint is associated with paclitaxel resistance in ovarian cancer. J. Ovarian Res. 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavallaris, M.; Kuo, D.Y.S.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific β-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.B.; Lothstein, L.; Manfredi, J.J.; Mellado, W.; Parness, J.; Roy, S.N.; Schiff, P.B.; Sorbara, L.; Zeheb, R. Taxol: Mechanisms of Action and Resistance. Ann. New York Acad. Sci. 1986, 466, 733–744. [Google Scholar] [CrossRef]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Blázquez, A.; González, M.; Álvarez, R.; del Mazo, S.; Medarde, M.; Peláez, R. Antitubulin sulfonamides: The successful combination of an established drug class and a multifaceted target. Med. Res. Rev. 2019, 39, 775–830. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Huang, Y.-W.; Yang, C.-H.; Huang, K.-S. Drug Delivery Systems and Combination Therapy by Using Vinca Alkaloids. Curr. Top. Med. Chem. 2015, 15, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, R.; Aramburu, L.; Gajate, C.; Vicente-Blázquez, A.; Mollinedo, F.; Medarde, M.; Peláez, R. Potent colchicine-site ligands with improved intrinsic solubility by replacement of the 3,4,5-trimethoxyphenyl ring with a 2-methylsulfanyl-6-methoxypyridine ring. Bioorg. Chem. 2020, 98, 103755. [Google Scholar] [CrossRef]
- Lee, R.M.; Gewirtz, D.A. Colchicine site inhibitors of microtubule integrity as vascular disrupting agents. Drug Dev. Res. 2008, 69, 352–358. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Zelcer, N.; Allen, J.D.; Yao, D.; Boyd, G.; Maliepaard, M.; Friedberg, T.H.; Smyth, J.F.; Jodrell, D.I. Glucuronidation as a mechanism of intrinsic drug resistance in colon cancer cells: Contribution of drug transport proteins. Biochem. Pharmacol. 2004, 67, 31–39. [Google Scholar] [CrossRef] [PubMed]
- ABT-751 ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 21 May 2020).
- Seddigi, Z.S.; Malik, M.S.; Saraswati, A.P.; Ahmed, S.A.; Babalghith, A.O.; Lamfon, H.A.; Kamal, A. Recent advances in combretastatin based derivatives and prodrugs as antimitotic agents. Medchemcomm 2017, 8, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Pandeya, S.; Kumar, P.; Sharma, P.; Gupta, S.; Soni, N.; Verma, K.; Bhardwaj, G. Combretastatin A-4 Analogs as Anticancer Agents. Mini Rev. Med. Chem. 2007, 7, 1186–1205. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, J.E.; Cook, J.A.; Lipschultz, C.; Teague, D.; Fisher, J.; Mitchell, J.B. Cytotoxic studies of paclitaxel (Taxol®) in human tumour cell lines. Br. J. Cancer 1993, 68, 1104–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naret, T.; Khelifi, I.; Provot, O.; Bignon, J.; Levaique, H.; Dubois, J.; Souce, M.; Kasselouri, A.; Deroussent, A.; Paci, A.; et al. 1,1-Diheterocyclic Ethylenes Derived from Quinaldine and Carbazole as New Tubulin-Polymerization Inhibitors: Synthesis, Metabolism, and Biological Evaluation. J. Med. Chem. 2019, 62, 1902–1916. [Google Scholar] [CrossRef]
- Alvarez, R.; Medarde, M.; Pelaez, R. New Ligands of the Tubulin Colchicine Site Based on X-Ray Structures. Curr. Top. Med. Chem. 2014, 14, 2231–2252. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Zardecki, C.; Di Costanzo, L.; Duarte, J.M.; Hudson, B.P.; Persikova, I.; Segura, J.; Shao, C.; Voigt, M.; Westbrook, J.D.; et al. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci. 2020, 29, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.L.; McGrath, C.; Hermone, A.R.; Burnett, J.C.; Zaharevitz, D.W.; Day, B.W.; Wipf, P.; Hamel, E.; Gussio, R. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J. Med. Chem. 2005, 48, 6107–6116. [Google Scholar] [CrossRef] [PubMed]
- Schobert, R.; Effenberger-Neidnicht, K.; Biersack, B. Stable combretastatin A-4 analogues with sub-nanomolar efficacy against chemoresistant HT-29 cells. Int. J. Clin. Pharmacol. Ther. 2011, 49, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.J.; Lin, S.S.; Wu, W.R.; Chen, L.R.; Li, C.F.; De Chen, H.; Chou, C.T.; Chen, Y.C.; Liang, S.S.; Chien, S.T.; et al. A microtubule inhibitor, ABT-751, induces autophagy and delays apoptosis in Huh-7 cells. Toxicol. Appl. Pharmacol. 2016, 311, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kanthou, C.; Greco, O.; Stratford, A.; Cook, I.; Knight, R.; Benzakour, O.; Tozer, G. The tubulin-binding agent combretastatin A-4-phosphate arrests endothelial cells in mitosis and induces mitotic cell death. Am. J. Pathol. 2004, 165, 1401–1411. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.H.; Sladek, T.L.; Liu, X.; Butler, B.R.; Froelich, C.J.; Thor, A.D. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001, 61, 348–354. [Google Scholar] [PubMed]
- Walker, P.R.; Leblanc, J.; Carson, C.; Ribecco, M.; Sikorska, M. Neither Caspase-3 nor DNA Fragmentation Factor Is Required for High Molecular Weight DNA Degradation in Apoptosis. Ann. N. Y. Acad. Sci. 1999, 887, 48–59. [Google Scholar] [CrossRef]
- Kagawa, S.; Gu, J.; Honda, T.; McDonnell, T.J.; Swisher, S.G.; Roth, J.A.; Fang, B. Deficiency of caspase-3 in MCF7 cells blocks Bax-mediated nuclear fragmentation but not cell death. Clin. Cancer Res. 2001, 7, 1474–1480. [Google Scholar] [PubMed]
- Stefański, T.; Mikstacka, R.; Kurczab, R.; Dutkiewicz, Z.; Kucińska, M.; Murias, M.; Zielińska-Przyjemska, M.; Cichocki, M.; Teubert, A.; Kaczmarek, M.; et al. Design, synthesis, and biological evaluation of novel combretastatin A-4 thio derivatives as microtubule targeting agents. Eur. J. Med. Chem. 2018, 144, 797–816. [Google Scholar] [CrossRef]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, M.; Ellahioui, Y.; Álvarez, R.; Gallego-Yerga, L.; Caballero, E.; Vicente-Blázquez, A.; Ramudo, L.; Marín, M.; Sanz, C.; Medarde, M.; et al. The Masked Polar Group Incorporation (MPGI) Strategy in Drug Design: Effects of Nitrogen Substitutions on Combretastatin and Isocombretastatin Tubulin Inhibitors. Molecules 2019, 24, 4319. [Google Scholar] [CrossRef] [Green Version]
- Shelanski, M.L.; Gaskin, F.; Cantor, C.R. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. USA 1973, 70, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Dumortier, C.; Gorbunoff, M.J.; Andreu, J.M.; Engelborghs, Y. Different Kinetic Pathways of the Binding of Two Biphenyl Analogues of Colchicine to Tubulin. Biochemistry 1996, 35, 4387–4395. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2007; pp. 319–326. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvin 17.8 ChemAxon. Available online: https://www.chemaxon.com (accessed on 2 May 2020).
- OpenEye Scientific Software, Inc. Santa Fe. Available online: https://www.eyesopen.com/ (accessed on 2 May 2020).
- Garcia-Perez, C.; Pelaez, R.; Theron, R.; Luis Lopez-Perez, J. JADOPPT: Java based AutoDock preparing and processing tool. Bioinformatics 2017, 33, 583–585. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Series 1: 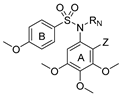 | Antiproliferative Activity IC50 (nM) | TPI | ||||
| TPI % | ||||||
| Z | RN | Compound | HeLa | MCF7 | 10 µM | IC50 (µM) |
| H | H | 1a | 240 | 375 | 0 | >20 |
| H | SO2–4-OMePh | 1b | >1000 | >1000 | 0 | >20 |
| H | Me | 2a | 71 | 127 | 35 | >20 |
| H | CH2-Dim | 2b | >1000 | >1000 | 0 | >20 |
| H | Et | 3 | 99 | 87 | 25 | >20 |
| H | EtBr | 4 | 94 | 340 | 30 | >20 |
| H | Ac | 5 | 287 | 270 | 0 | >20 |
| H | CH2CN | 6 | 143 | 275 | 0 | >20 |
| H | CH2COOEt | 7 | 217 | 335 | 21 | >20 |
| H | CH2COOH | 8 | >1000 | >1000 | 0 | >20 |
| H | Benzyl | 9 | 750 | 830 | 21 | >20 |
| H | COOBenzyl | 10 | >1000 | >1000 | 11 | >20 |
| NO2 | H | 11 | >1000 | >1000 | 10 | >20 |
| NH2 | H | 12 | >1000 | >1000 | 0 | >20 |
| NH2 | Me | 13 | >1000 | >1000 | 0 | >20 |
| NH2 | CH2-Dim | 14 | >1000 | >1000 | 0 | >20 |
| NHAc | H | 15 | >1000 | >1000 | 0 | >20 |
| NHAc | Me | 16 | >1000 | >1000 | 0 | >20 |
| NHCHO | H | 17 | >1000 | >1000 | 4 | >20 |
| N(CH3)2 | H | 18 | >1000 | >1000 | 0 | >20 |
| N(CH3)2 | Me | 19 | >1000 | >1000 | 0 | >20 |
| Br | H | 20 | >1000 | >1000 | 0 | >20 |
| Gly-tBOC | H | 21 | >1000 | >1000 | 0 | >20 |
| Succinic | H | 22 | >1000 | >1000 | 0 | >20 |
| -N=N- | 23 | >1000 | >1000 | 0 | >20 | |
| -N=CH- | 24 | >1000 | >1000 | 0 | >20 | |
Series 2:  | Antiproliferative Activity IC50 (nM) | TPI | ||||
| TPI % | ||||||
| R | RN | Compound | HeLa | MCF7 | 10 µM | IC50 (µM) |
| NO2 | H | 25 | >1000 | >1000 | 0 | >20 |
| NH2 | H | 26 | >1000 | >1000 | 0 | >20 |
| NH2 | Me | 27 | >1000 | >1000 | 0 | >20 |
| N(CH3)2 | H | 28 | 230 | 173 | 0 | >20 |
| N(CH3)2 | Me | 29a | 63 | 30 | 40 | >20 |
| N(CH3)2 | CH2-Dim | 29b | >1000 | >1000 | 0 | >20 |
| N(CH3)2 | Et | 30 | 66 | 81 | 45 | >20 |
| N(CH3)2 | Ac | 31 | 183 | 135 | 38 | 14 |
| N(CH3)2 | CH2CN | 32 | 55 | 120 | 17 | >20 |
| N(CH3)2 | CH2COOEt | 33 | 99 | 91 | 14 | >20 |
| N(CH3)2 | CH2COOH | 34 | >1000 | >1000 | 6 | >20 |
| N(CH3)2 | Benzyl | 35a | 330 | 100 | 72 | 5.3 |
| N(CH3)2 | COOBenzyl | 35b | >1000 | >1000 | 0 | >20 |
| NHCHO | H | 36 | >1000 | >1000 | 0 | >20 |
| NHCH3 | H | 37 | 607 | >1000 | 7 | >20 |
| NHCH3 | Me | 38 | 44 | 61 | 47 | 10 |
Series 3: 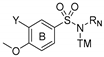 | Antiproliferative Activity IC50 (nM) | TPI | ||||
| TPI % | ||||||
| Y | RN | Compound | HeLa | MCF7 | 10 µM | IC50 (µM) |
| NO2 | H | 40 | >1000 | >1000 | 0 | >20 |
| NH2 | H | 41 | 260 | 96 | 15 | >20 |
| NH2 | Me | 42 | 23 | 26 | 41 | 12 |
| NH2 | Et | 43 | 38 | 14 | 37 | >20 |
| NH2 | CH2CN | 44 | 60 | 8 | 5 | >20 |
| NH2 | Benzyl | 45 | 25 | 48 | 75 | 3.7 |
| NHCH2COOEt | Me | 46 | 210 | 48 | 13 | >20 |
| NHCH2COOEt | CH2COOEt | 47 | >1000 | >1000 | 0 | >20 |
| NHCH3 | Me | 48a | 440 | 1190 | 2 | >20 |
| N(CH3)2 | Me | 48b | >1000 | >1000 | 0 | >20 |
| Paclitaxel [52] | 2.6 | 2.5 | n.d. 1 | n.d. | ||
| Combretastatin A-4 | 2 | 1 | 100 | 3 | ||
| ABT-751 | 388 | 180 | 69 | 4.4 | ||
| Compound | Solub (µg/mL) | Compound | Solub (µg/mL) | Compound | Solub (µg/mL) |
|---|---|---|---|---|---|
| 1a | 83 | 22 | 1690 | 37 | 43 |
| 1b | 108 | 23 | 41 | 38 | 16 |
| 2a | 27 | 26 | 28 | 41 | 89 |
| 3 | 30 | 27 | 25 | 42 | 108 |
| 5 | 38 | 28 | 15 | 43 | 58 |
| 6 | 33 | 29a | 7 | 44 | 46 |
| 11 | 88 | 29b | 4 | 45 | 14 |
| 12 | 158 | 30 | 6 | CA-4 | 1 |
| 15 | 230 | 31 | 18 | ABT-751 | 40 |
| 20 | 87 | 32 | 8 | Paclitaxel [23,24,25] | <2 |
| 21 | 357 | 36 | 27 | – | – |
| Antiproliferative Activity IC50 (nM) | |||||
|---|---|---|---|---|---|
| Compound | SKOV3 | IGROV-1 | A2780 | OVCAR-8 | OVCAR-3 |
| 38 | 46 | 248 | 68 | 74 | 31 |
| 42 | 7 | 400 | 42 | 37 | 72 |
| 45 | 48 | 492 | 104 | 48 | 58 |
| Paclitaxel1 | 81 | 39 | 3 | 6 | 17 |
| Antiproliferative Activity IC50 (nM) | ||
|---|---|---|
| Compound | HT-29 | HT-29 Verapamil 10 µM |
| 38 | 59 | 50 |
| 42 | 81 | 79 |
| 45 | 300 | 276 |
| CA-4 | 305 | 327 |
| Compound | Annexin V-FITC/PI 72 h (%) | Annexin V-FITC/PI 6 Days (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Live | EA 1 | LA 2 | Necrosis | Live | EA | LA | Necrosis | ||
| MCF7 | 38 (600 nM) | 60.5 | 29.0 | 5.4 | 5.1 | 52.0 | 45.0 | 2.7 | 0.3 |
| 42 (50 nM) | 87.7 | 8.5 | 2.3 | 1.4 | 79.1 | 18.9 | 1.7 | 0.3 | |
| 45 (400 nM) | 70.5 | 20.1 | 7.5 | 1.9 | 56.7 | 39.3 | 3.8 | 0.3 | |
| CA-4 (50 nM) | 58.3 | 31.7 | 3.3 | 6.7 | 44.5 | 52.4 | 2.7 | 0.4 | |
| Control | 93.6 | 1.8 | 2.9 | 1.7 | 97.4 | 0.2 | 0.8 | 1.6 | |
| SKOV3 | 38 (600 nM) | 62.3 | 30.5 | 5.4 | 1.8 | ||||
| 42 (50 nM) | 85.1 | 12.2 | 2.0 | 0.7 | |||||
| 45 (400 nM) | 72.7 | 23.4 | 2.6 | 1.3 | |||||
| Paclitaxel (10 nM) | 90.7 | 3.8 | 3.1 | 2.4 | |||||
| Control | 99.2 | 0.5 | 0.3 | 0.1 | |||||
| HeLa | 38 (600 nM) | 0.6 | 1.1 | 87.6 | 10.6 | ||||
| 42 (50 nM) | 7.3 | 1.4 | 82.0 | 9.4 | |||||
| 45 (400 nM) | 0.8 | 1.5 | 84.9 | 12.7 | |||||
| Control | 90.5 | 1.7 | 5.5 | 2.4 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, M.; Ovejero-Sánchez, M.; Vicente-Blázquez, A.; Álvarez, R.; Herrero, A.B.; Medarde, M.; González-Sarmiento, R.; Peláez, R. Microtubule Destabilizing Sulfonamides as an Alternative to Taxane-Based Chemotherapy. Int. J. Mol. Sci. 2021, 22, 1907. https://doi.org/10.3390/ijms22041907
González M, Ovejero-Sánchez M, Vicente-Blázquez A, Álvarez R, Herrero AB, Medarde M, González-Sarmiento R, Peláez R. Microtubule Destabilizing Sulfonamides as an Alternative to Taxane-Based Chemotherapy. International Journal of Molecular Sciences. 2021; 22(4):1907. https://doi.org/10.3390/ijms22041907
Chicago/Turabian StyleGonzález, Myriam, María Ovejero-Sánchez, Alba Vicente-Blázquez, Raquel Álvarez, Ana B. Herrero, Manuel Medarde, Rogelio González-Sarmiento, and Rafael Peláez. 2021. "Microtubule Destabilizing Sulfonamides as an Alternative to Taxane-Based Chemotherapy" International Journal of Molecular Sciences 22, no. 4: 1907. https://doi.org/10.3390/ijms22041907
APA StyleGonzález, M., Ovejero-Sánchez, M., Vicente-Blázquez, A., Álvarez, R., Herrero, A. B., Medarde, M., González-Sarmiento, R., & Peláez, R. (2021). Microtubule Destabilizing Sulfonamides as an Alternative to Taxane-Based Chemotherapy. International Journal of Molecular Sciences, 22(4), 1907. https://doi.org/10.3390/ijms22041907