
Computational Modulation of the V3 Region of Glycoprotein gp125 of HIV-2




Abstract
:1. Introduction
2. Results
2.1. Modeling the Structure of HIV-2 V3 Loop
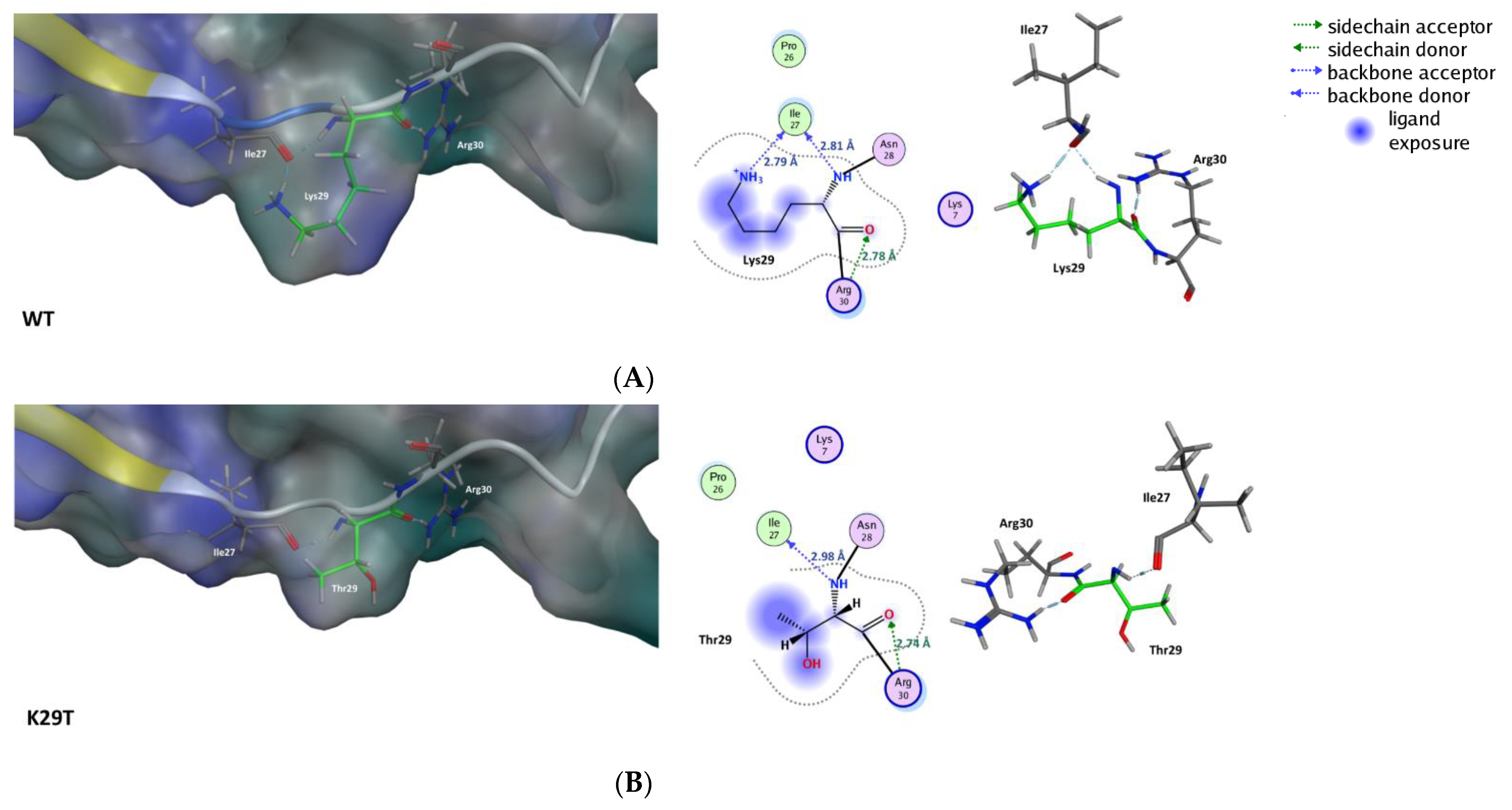
2.2. Structural Analysis of WT and Mutated HIV-2 V3 Loop
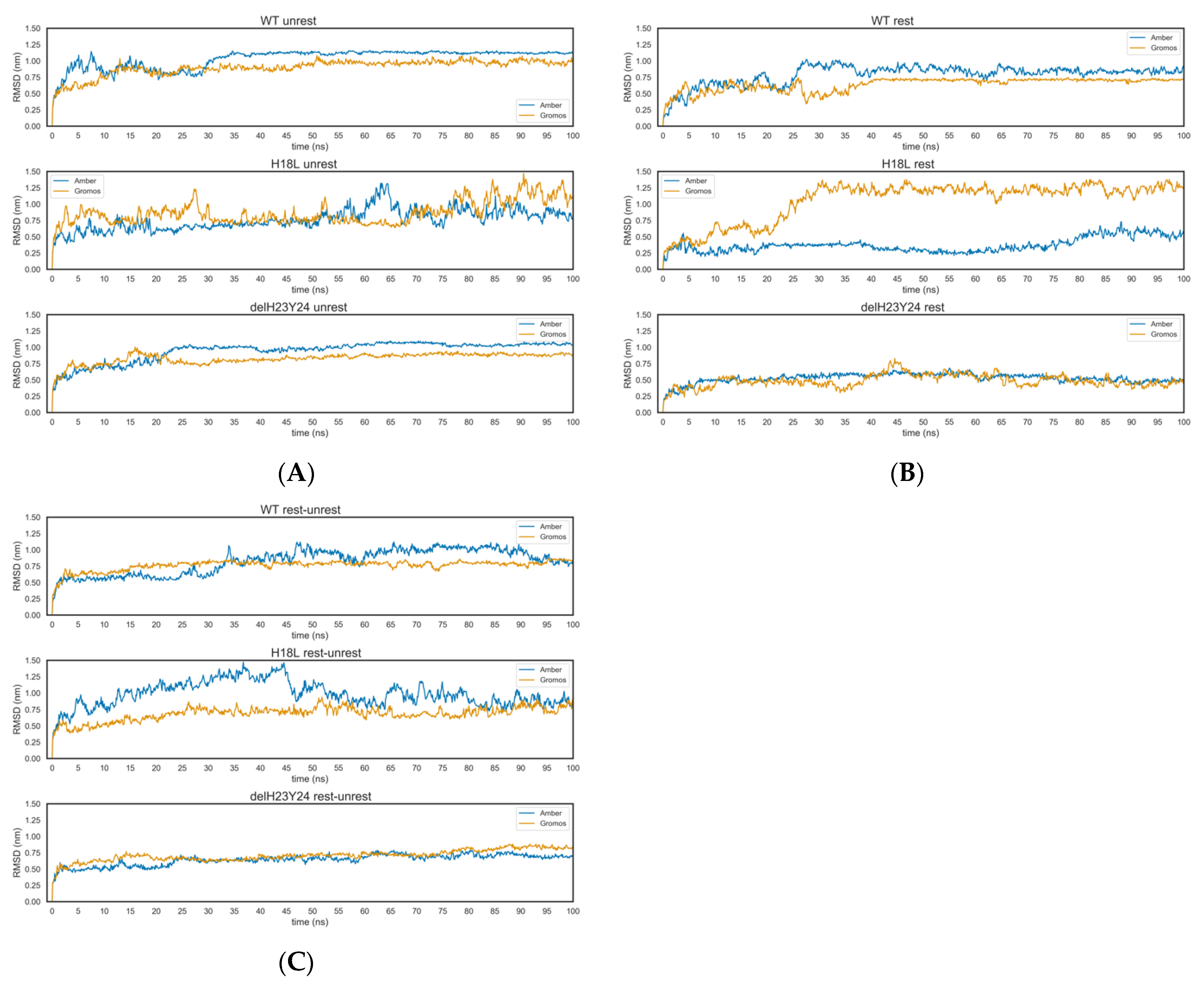
2.3. Molecular Dynamics
3. Discussion
Molecular Interactions in the MD Runs
4. Materials and Methods
4.1. Modelling the Structure of HIV-2ROD V3 Region
4.2. Molecular Dynamics of the Constructed Models
4.2.1. System Setup
4.2.2. Simulation Details
4.3. Key Interaction Determination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ekouevi, D.K.; Balestre, E.; Coffie, P.A.; Minta, D.; Messou, E.; Sawadogo, A.; Minga, A.; Sow, P.S.; Bissagnene, E.; Eholie, S.P.; et al. Characteristics of HIV-2 and HIV-1/HIV-2 Dually Seropositive Adults in West Africa Presenting for Care and Antiretroviral Therapy: The IeDEA-West Africa HIV-2 Cohort Study. PLoS ONE 2013, 8, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, G.S.; Raugi, D.N.; Smith, R.A. 90-90-90 for HIV-2? Ending the HIV-2 epidemic by enhancing care and clinical management of patients infected with HIV-2. Lancet HIV 2018, 5, e390–e399. [Google Scholar] [CrossRef]
- Nyamweya, S.; Hegedus, A.; Jaye, A.; Rowland-Jones, S.; Flanagan, K.L.; Macallan, D.C. Comparing HIV-1 and HIV-2 infection: Lessons for viral immunopathogenesis. Rev. Med. Virol. 2013, 23, 221–240. [Google Scholar] [CrossRef]
- Joint United Nations Program on HIV/AIDS To Help End the AIDS Epidemic. 2014.
- Hønge, B.L.; Petersen, M.S.; Jespersen, S.; Medina, C.; Té, D.D.S.; Kjerulff, B.; Engell-Sørensen, T.; Madsen, T.; Laursen, A.L.; Wejse, C.; et al. T-cell and B-cell perturbations identify distinct differences in HIV-2 compared with HIV-1-induced immunodeficiency. Aids 2019, 33, 1131–1141. [Google Scholar] [CrossRef]
- Tendeiro, R.; Fernandes, S.; Foxall, R.B.; Marcelino, J.M.; Taveira, N.; Soares, R.S.; Baptista, A.P.; Cavaleiro, R.; Gomes, P.; Victorino, R.M.M.; et al. Memory B-cell depletion is a feature of HIV-2 infection even in the absence of detectable viremia. Aids 2012, 26, 1607–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchtenhagen, H.; Friemann, R.; Raszewski, G.; Spetz, A.-L.; Nilsson, L.; Achour, A. Crystal structure of the HIV-2 neutralizing Fab fragment 7C8 with high specificity to the V3 region of gp125. PLoS ONE 2011, 6, e18767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego, P.; Calado, R.; Marcelino, J.M.; Pereira, P.; Quintas, A.; Barroso, H.; Taveira, N. An ancestral HIV-2/simian immunodeficiency virus peptide with potent HIV-1 and HIV-2 fusion inhibitor activity. Aids 2013, 27, 1081–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Silva, T.I.; Cotten, M.; Rowland-Jones, S.L. HIV-2: The forgotten AIDS virus. Trends Microbiol. 2008, 16, 588–595. [Google Scholar] [CrossRef]
- Campbell-Yesufu, O.T.; Gandhi, R.T. Update on human immunodeficiency virus (HIV)-2 infection. Clin. Infect. Dis. 2011, 52, 780–787. [Google Scholar] [CrossRef] [Green Version]
- Marcelino, J.M.; Borrego, P.; Nilsson, C.; Família, C.; Barroso, H.; Maltez, F.; Doroana, M.; Antunes, F.; Quintas, A.; Taveira, N. Resistance to antibody neutralization in HIV-2 infection occurs in late stage disease and is associated with X4 tropism. Aids 2012, 26, 2275–2284. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.; Calado, R.; Borrego, P.; Marcelino, J.M.; Bártolo, I.; Rosado, L.; Cavaco-Silva, P.; Gomes, P.; Família, C.; Quintas, A.; et al. Evolution of the human immunodeficiency virus type 2 envelope in the first years of infection is associated with the dynamics of the neutralizing antibody response. Retrovirology 2013, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esbjörnsson, J.; Månsson, F.; Kvist, A.; da Silva, Z.J.; Andersson, S.; Fenyö, E.M.; Isberg, P.E.; Biague, A.J.; Lindman, J.; Palm, A.A.; et al. Long-term follow-up of HIV-2-related AIDS and mortality in Guinea-Bissau: A prospective open cohort study. Lancet HIV 2019, 6, e25–e31. [Google Scholar] [CrossRef]
- Tzou, P.L.; Descamps, D.; Rhee, S.-Y.; Raugi, D.N.; Charpentier, C.; Taveira, N.; Smith, R.A.; Soriano, V.; de Mendoza, C.; Holmes, S.P.; et al. Expanded Spectrum of Antiretroviral-Selected Mutations in Human Immunodeficiency Virus Type 2. J. Infect. Dis. 2020, 221, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Vogan, E.M.; Gong, H.; Skehel, J.J.; Wiley, D.C.; Harrison, S.C. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 2005, 433, 834–841. [Google Scholar] [CrossRef]
- Teixeira, C.; Gomes, J.R.; Gomes, P.; Maurel, F.; Barbault, F. Viral surface glycoproteins, gp120 and gp41, as potential drug targets against HIV-1: Brief overview one quarter of a century past the approval of zidovudine, the first anti-retroviral drug. Eur. J. Med. Chem. 2011, 46, 979–992. [Google Scholar] [CrossRef] [Green Version]
- LaLonde, J.M.; Kwon, Y.D.; Jones, D.M.; Sun, A.W.; Courter, J.R.; Soeta, T.; Kobayashi, T.; Princiotto, A.M.; Wu, X.; Schön, A.; et al. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J. Med. Chem. 2012, 55, 4382–4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Borrego, P.; Ding, X.; Zhu, Y.; Martins, A.; Chong, H.; Taveira, N.; He, Y. A Helical Short-Peptide Fusion Inhibitor with Highly Potent Activity against Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol. 2017, 91, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.; Bettadapura, R.; Bajaj, C. X-ray, Cryo-EM, and computationally predicted protein structures used in integrative modeling of HIV Env glycoprotein gp120 in complex with CD4 and 17b. Data Br. 2016, 6, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Kadow, J.F.; Sivaprakasam, P.; Meanwell, N.A.; Roy Kimura, S.; Dicker, I.; Wang, T.; Zhou, N.; Krystal, M.; Langley, D.R.; McAuliffe, B. Homology models of the HIV-1 attachment inhibitor BMS-626529 bound to gp120 suggest a unique mechanism of action. Proteins Struct. Funct. Bioinform. 2014, 83, 331–350. [Google Scholar]
- Clark, A.J.; Gindin, T.; Zhang, B.; Wang, L.; Abel, R.; Murret, C.S.; Xu, F.; Bao, A.; Lu, N.J.; Zhou, T.; et al. Free Energy Perturbation Calculation of Relative Binding Free Energy between Broadly Neutralizing Antibodies and the gp120 Glycoprotein of HIV-1. J. Mol. Biol. 2017, 429, 930–947. [Google Scholar] [CrossRef]
- Visseaux, B.; Hurtado-Nedelec, M.; Charpentier, C.; Collin, G.; Storto, A.; Matheron, S.; Larrouy, L.; Damond, F.; Brun-Vezinet, F.; Descamps, D. Molecular Determinants of HIV-2 R5-X4 Tropism in the V3 Loop: Development of a New Genotypic Tool. J. Infect. Dis. 2012, 205, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Döring, M.; Borrego, P.; Büch, J.; Martins, A.; Friedrich, G.; Camacho, R.J.; Eberle, J.; Kaiser, R.; Lengauer, T.; Taveira, N.; et al. A genotypic method for determining HIV-2 coreceptor usage enables epidemiological studies and clinical decision support. Retrovirology 2016, 13, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges-Mameletzis, I.; De Bree, G.J.; Rowland-Jones, S.L. An underestimated lentivirus model: What can HIV-2 research contribute to the development of an effective HIV-1 vaccine? Expert Rev. Anti. Infect. Ther. 2011, 9, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Davenport, Y.W.; West, A.P.; Bjorkman, P.J. Structure of an HIV-2 gp120 in Complex with CD4. J. Virol. 2016, 90, 2112–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE).
- Barroso, H.; Borrego, P.; Bártolo, I.; Marcelino, J.M.; Família, C.; Quintas, A.; Taveira, N. Evolutionary and Structural Features of the C2, V3 and C3 Envelope Regions Underlying the Differences in HIV-1 and HIV-2 Biology and Infection. PLoS ONE 2011, 6, e14548. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Lam, S.N.; Acharya, P.; Tang, M.; Xiang, S.H.; Hussan, S.S.; Stanfield, R.L.; Robinson, J.; Sodroski, J.; Wilson, I.A.; et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 2007, 317, 1930–1934. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-C. Structure of a V3-Containing HIV-1 gp120 Core. Science 2005, 310, 1025–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, F.; Sánchez, R.; Sali, A. Statistical potentials for fold assessment. Protein Sci. 2009, 11, 430–448. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Narumi, T.; Arai, H.; Yoshimura, K.; Harada, S.; Nomura, W.; Matsushita, S.; Tamamura, H. Small molecular CD4 mimics as HIV entry inhibitors. Bioorganic Med. Chem. 2011, 19, 6735–6742. [Google Scholar] [CrossRef]
- Liao, S.M.; Du, Q.S.; Meng, J.Z.; Pang, Z.W.; Huang, R.B. The multiple roles of histidine in protein interactions. Chem. Cent. J. 2013, 7, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Madani, N.; Princiotto, A.M.; Zhao, C.; Jahanbakhshsefidi, F.; Mertens, M.; Herschhorn, A.; Melillo, B.; Smith, A.B., III; Sodroski, J. Activation and Inactivation of Primary Human Immunodeficiency Virus Envelope Glycoprotein Trimers by CD4- Mimetic Compounds Navid. J. Virol. 2017, 91, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Santos-Costa, Q.; Parreira, R.; Moniz-Pereira, J.; Azevedo-Pereira, J.M. Molecular Characterization of the env Gene of Two CCR5/CXCR4-Independent Human Immunodeficiency 2 Primary Isolates. J. Med. Virol. 2009, 81, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zou, X. Modeling Protein Flexibility in Molecular Docking, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 3–8, ISBN 9780128032008. [Google Scholar]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, 349–357. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Poger, D.; Van Gunsteren, W.F.; Mark, A.E. A new force field for simulating phosphatidylcholine bilayers. J. Comput. Chem. 2010, 31, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Poger, D.; Mark, A.E. On the validation of molecular dynamics simulations of saturated and cis-monounsaturated phosphatidylcholine lipid bilayers: A comparison with experiment. J. Chem. Theory Comput. 2010, 6, 325–336. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101-1–014101-7. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.P.A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitztom, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Tamamis, P.; Floudas, C.A. Molecular recognition of CCR5 by an HIV-1 gp120 V3 loop. PLoS ONE 2014, 9, e95767. [Google Scholar] [CrossRef] [Green Version]
- Tamamis, P.; Floudas, C.A. Molecular recognition of CXCR4 by a dual tropic HIV-1 gp120 V3 loop. Biophys. J. 2013, 105, 1502–1514. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V3 Systems | V3 Sequence | |||||||
|---|---|---|---|---|---|---|---|---|
| ROD WT | 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 |
| CKRPGNKTVKQIMLMSGHVFHSHYQPINKRPRQAWC | ||||||||
| H18L | CKRPGNKTVKQIMLMSGLVFHSHYQPINKRPRQAWC | |||||||
| k29T | CKRPGNKTVKQIMLMSGHVFHSHYQPINTRPRQAWC | |||||||
| ΔH23Y24 | CKRPGNKTVKQIMLMSGHVFHSHYQPINKRPRQAWC | |||||||
| H18L_K29T | CKRPGNKTVKQIMLMSGLVFHSHYQPINTRPRQAWC | |||||||
| H18L_ΔH23Y24 | CKRPGNKTVKQIMLMSGLVFHSHYQPINKRPRQAWC | |||||||
| H18L_K29T_ΔH23Y24 | CKRPGNKTVKQIMLMSGLVFHSHYQPINTRPRQAWC | |||||||
| Strain of the Virus (PDB Identifier) | V3 Sequence | Res. (Å) |
|---|---|---|
| HIV-1YU2 (2QAD) | CTRPNNNTRKSINIQRGPGRALYTTGEIIGDIRQAHC | 3.3 |
| HIV-1 JR-FL (2B4C) | CTRPNQNTRKSIHIQRGPGRAFYTTGEIIGDIRQAHC | 3.5 |
| HIV-2ST (5CAY) | CKRPGNKTVVPITLMSGLVFHSHYQPINRRPRQAWC | 3.0 |
| HIV-2ROD | CKRPGNKTVKQIMLMSGHVFHSHYQPINKRPRQAWC | -- |
| V3 Sequence | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Residues | C | K | R | P | G | N | K | T | V | K | Q | I | M | L | M | S | G | H | V | F | H | S | H | Y | Q | P | I | N | K | R | P | R | Q | A | W | C |
| PSIPRED | ||||||||||||||||||||||||||||||||||||
| Robetta | ||||||||||||||||||||||||||||||||||||
| Model | ||||||||||||||||||||||||||||||||||||
| β Sheet | α Helix | |||||||||||||||||||||||||||||||||||
| V3 Region | RMSD | SASA (Å2) | Δ (SASA) | Charge | Length (Å) | Width (Å) |
|---|---|---|---|---|---|---|
| ROD WT | 0 | 3852.3 | 0 | +7 | 47.50 | 5.30 |
| H18L | 0.096 | 3853.1 | 0.8 | +7 | 47.67 | 5.34 |
| k29T | 0.144 | 3834.4 | −17.9 | +6 | 47.67 | 5.34 |
| ΔH23Y24 | 2.812 | 3790.8 | −61.5 | +7 | 47.67 | 6.17 |
| H18L_K29T | 0.150 | 3821.9 | −30.4 | +6 | 47.68 | 5.34 |
| H18L_ΔH23Y24 | 1.107 | 3778.5 | −73.8 | +7 | 47.50 | 6.13 |
| H18L_K29T_ΔH23Y24 | 1.128 | 3760.5 | −91.8 | +6 | 47.50 | 6.13 |
| V3 Region | Amber 99SB-ILDN | Gromos 54a7 | ||||
|---|---|---|---|---|---|---|
| ROD WT | C2 and C3 unrest | C2 and C3 rest | C2 and C3 unrest after restriction | C2 and C3 unrest | C2 and C3 rest | C2 and C3 unrest after restriction |
| H18L | C2 and C3 unrest | C2 and C3 rest | C2 and C3 unrest after restriction | C2 and C3 unrest | C2 and C3 rest | C2 and C3 unrest after restriction |
| ΔH23Y24 | C2 and C3 unrest | C2 and C3 rest | C2 and C3 unrest after restriction | C2 and C3 unrest | C2 and C3 rest | C2 and C3 unrest after restriction |
| Amber 99SB-ILDN | Gromos 54a7 | |||||
|---|---|---|---|---|---|---|
| Variant | Unrest | Rest | Unrest after Restriction | Unrest | Rest | Unrest after Restriction |
| ROD WT | −513,539.48 ± 1087.30 | −512,927.52 ± 1082.42 | −513,472.38 ± 1039.24 | −867,141.25 ± 1358.80 | −866,617.64 ± 1326.67 | −867,171.99 ± 1321.13 |
| H18L | −571,028.67 ± 1125.78 | −570,290.46 ± 1123.11 | −570,940.25 ± 1126.62 | −1,461,028.35 ± 1629.22 | −1,460,455.02 ± 1782.04 | −1,461,030.00 ± 1811,57 |
| ΔH23Y24 | −1,124,328.83 ± 1491.94 | −1,123,663.29 ± 1625.08 | −1,124,215.34 ± 1613.15 | −602,249.39 ± 1112.07 | −601,613.97 ± 1137.73 | −602,404.45 ± 1090.76 |
| Amber 99SB-ILDN | Gromos 54a7 | |||||
|---|---|---|---|---|---|---|
| Variant | Unrest | Rest | Unrest after Restriction | Unrest | Rest | Unrest after Restriction |
| ROD WT | 1.124 ± 0.015 | 0.849 ± 0.045 | 0.951 ± 0.087 | 0.966 ± 0.046 | 0.705 ± 0.018 | 0.789 ± 0.033 |
| H18L | 0.852 ± 0.132 | 0.393 ± 0.125 | 0.966 ± 0.149 | 0.911 ± 0.197 | 1.224 ± 0.065 | 0.729 ± 0.069 |
| ΔH23Y24 | 1.026 ± 0.041 | 0.542 ± 0.052 | 0.691 ± 0.041 | 0.870 ± 0.032 | 0.521 ± 0.091 | 0.754 ± 0.058 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, P.A.; Taveira, N.; Guedes, R.C. Computational Modulation of the V3 Region of Glycoprotein gp125 of HIV-2. Int. J. Mol. Sci. 2021, 22, 1948. https://doi.org/10.3390/ijms22041948
Serra PA, Taveira N, Guedes RC. Computational Modulation of the V3 Region of Glycoprotein gp125 of HIV-2. International Journal of Molecular Sciences. 2021; 22(4):1948. https://doi.org/10.3390/ijms22041948
Chicago/Turabian StyleSerra, Patrícia A., Nuno Taveira, and Rita C. Guedes. 2021. "Computational Modulation of the V3 Region of Glycoprotein gp125 of HIV-2" International Journal of Molecular Sciences 22, no. 4: 1948. https://doi.org/10.3390/ijms22041948
APA StyleSerra, P. A., Taveira, N., & Guedes, R. C. (2021). Computational Modulation of the V3 Region of Glycoprotein gp125 of HIV-2. International Journal of Molecular Sciences, 22(4), 1948. https://doi.org/10.3390/ijms22041948