
Binding of Amyloid β(1–42)-Calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
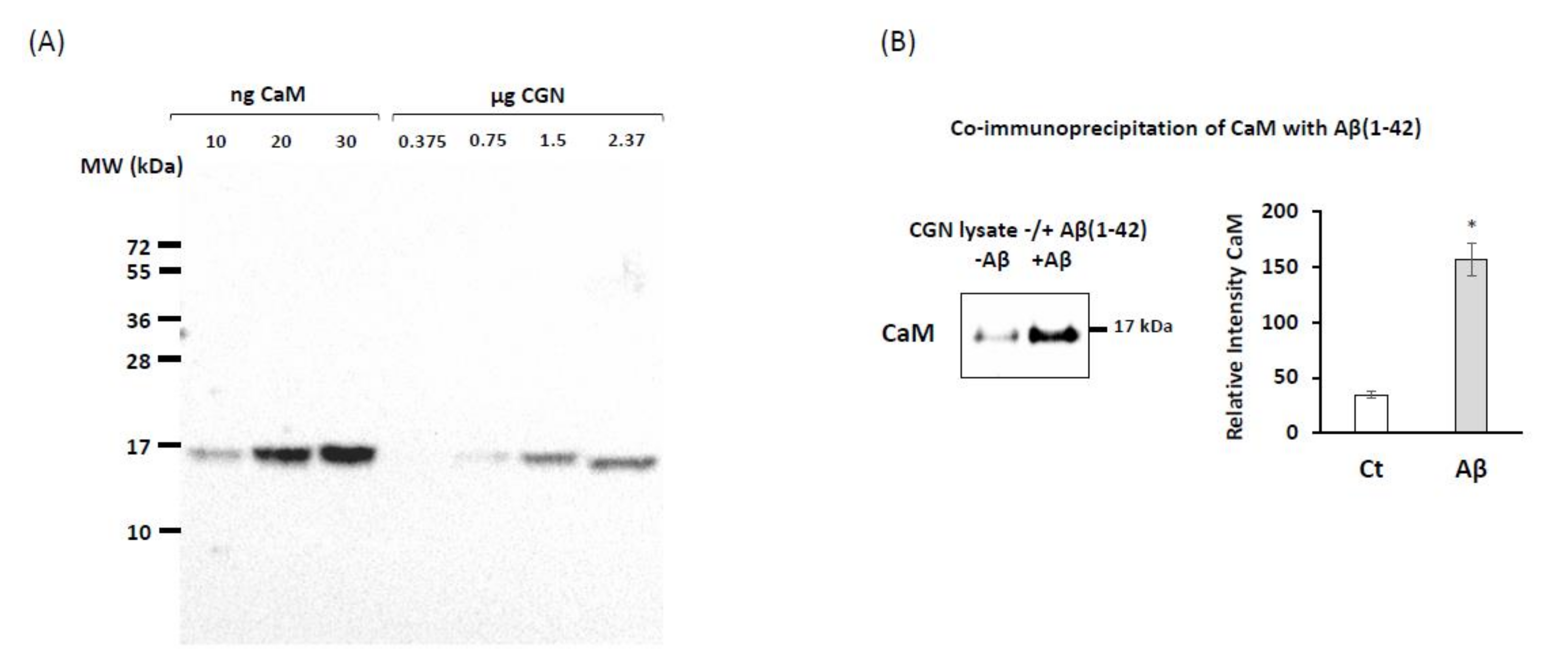
2.1. Co-Immunoprecipitation of Calmodulin (CaM) with Submicromolar Concentrations of Aβ(1–42) in Cerebellar Granule Neurons (CGN) Lysates
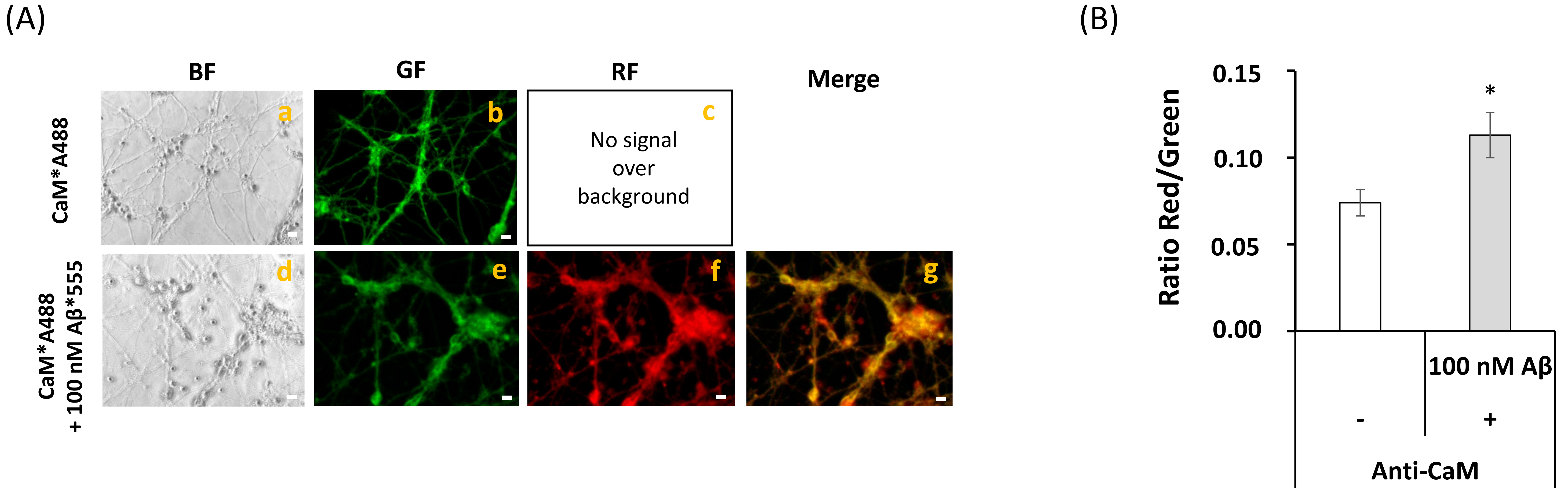
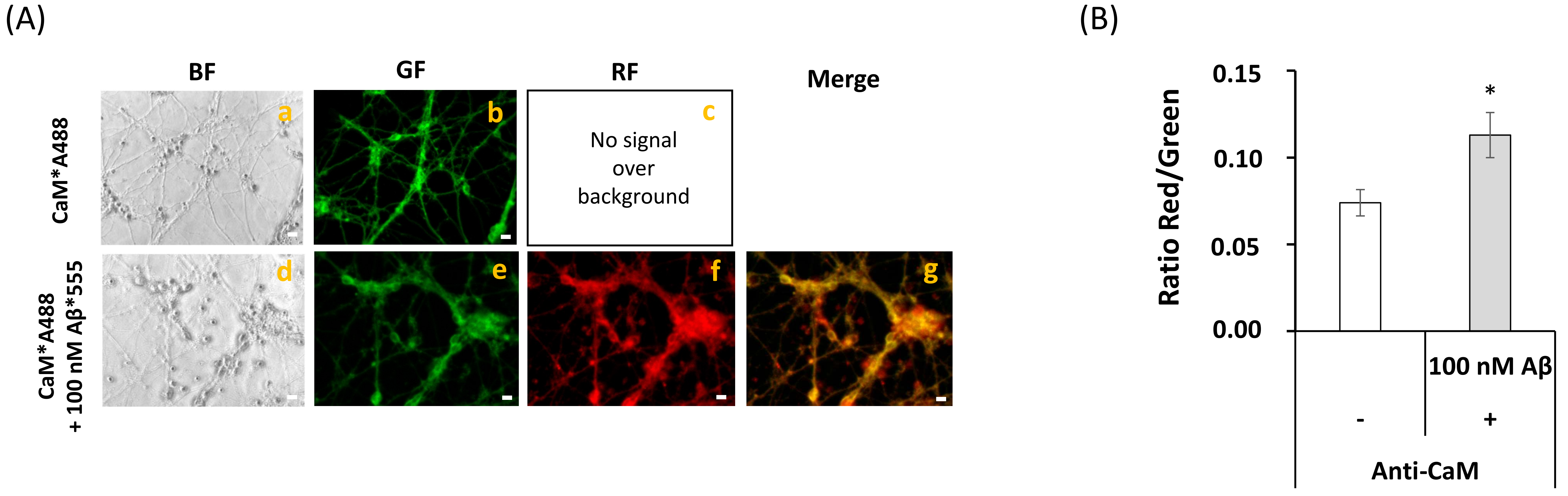
2.2. Fluorescence Resonance Energy Transfer (FRET) Imaging Shows an Extensive Co-Localization between CaM and Aβ(1–42) HiLyteTM-Fluor555 in Mature CGN
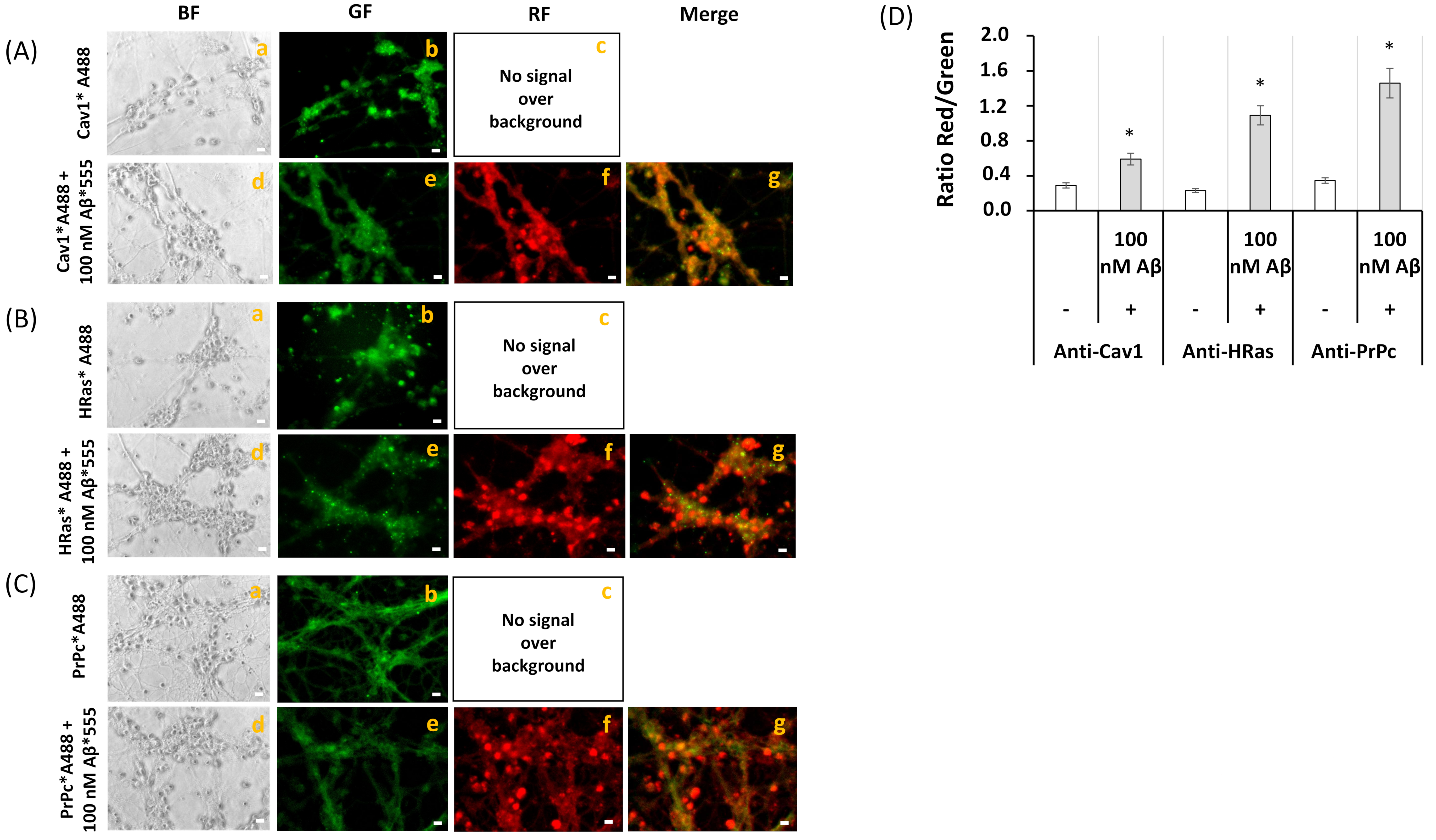
2.3. FRET Imaging Highlights the Association of Aβ(1–42)-HiLyteTM-Fluor555 and CaM with Lipid Rafts Markers in Mature CGN
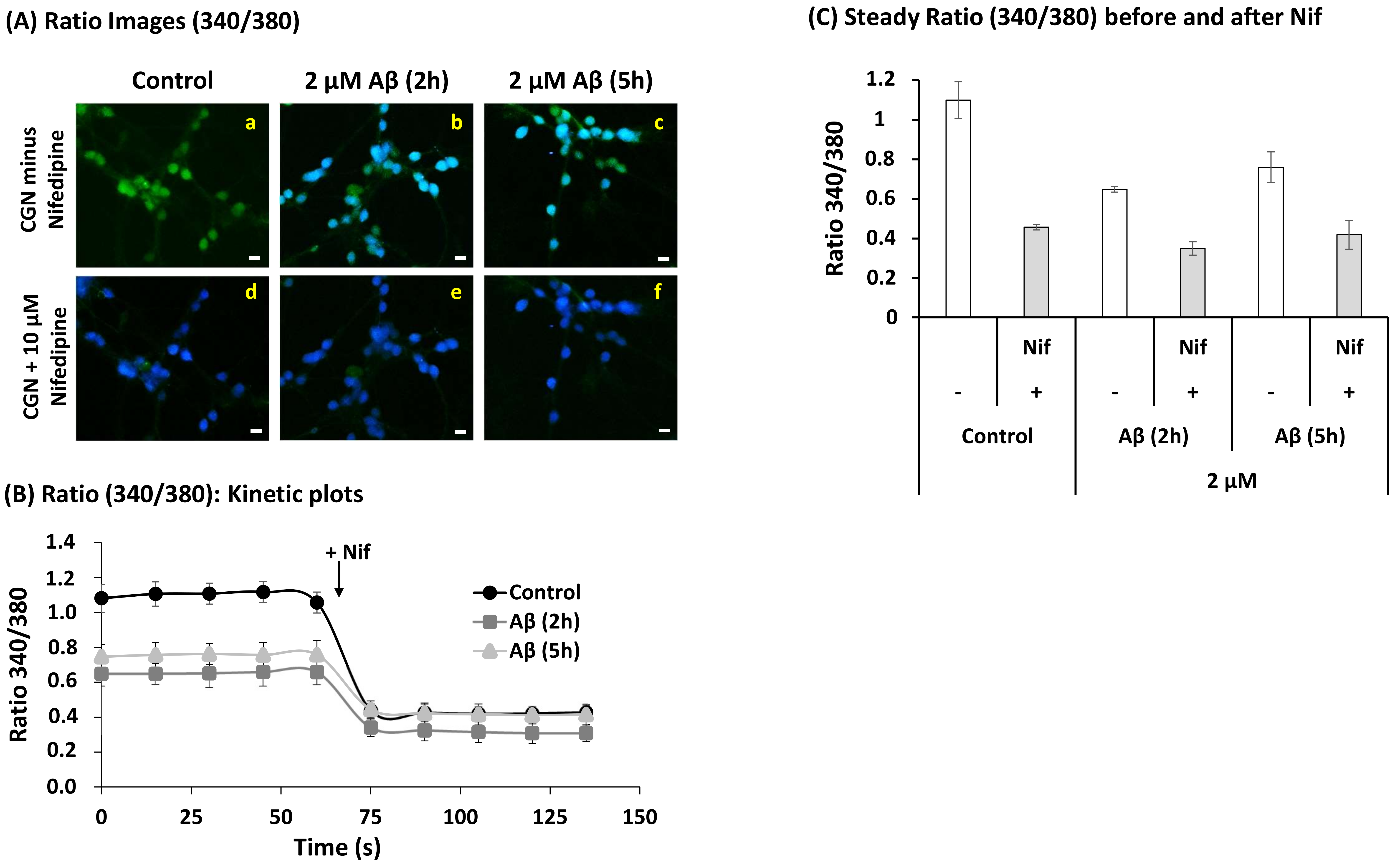
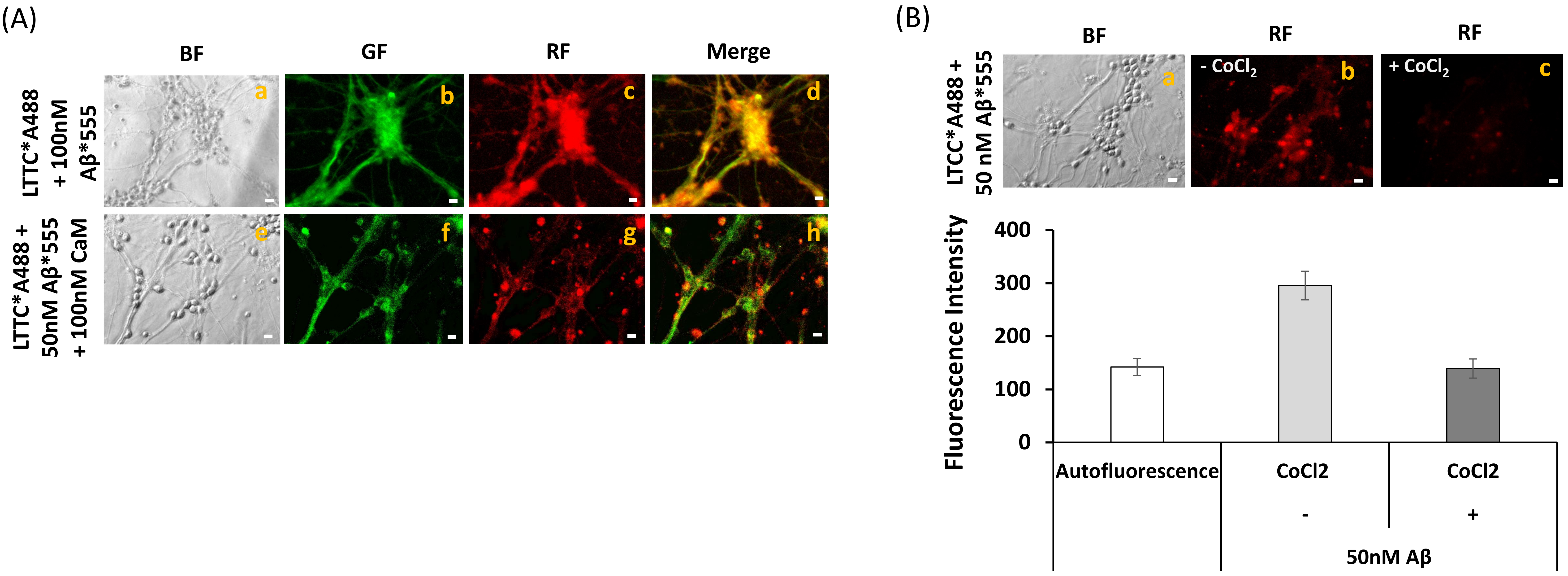
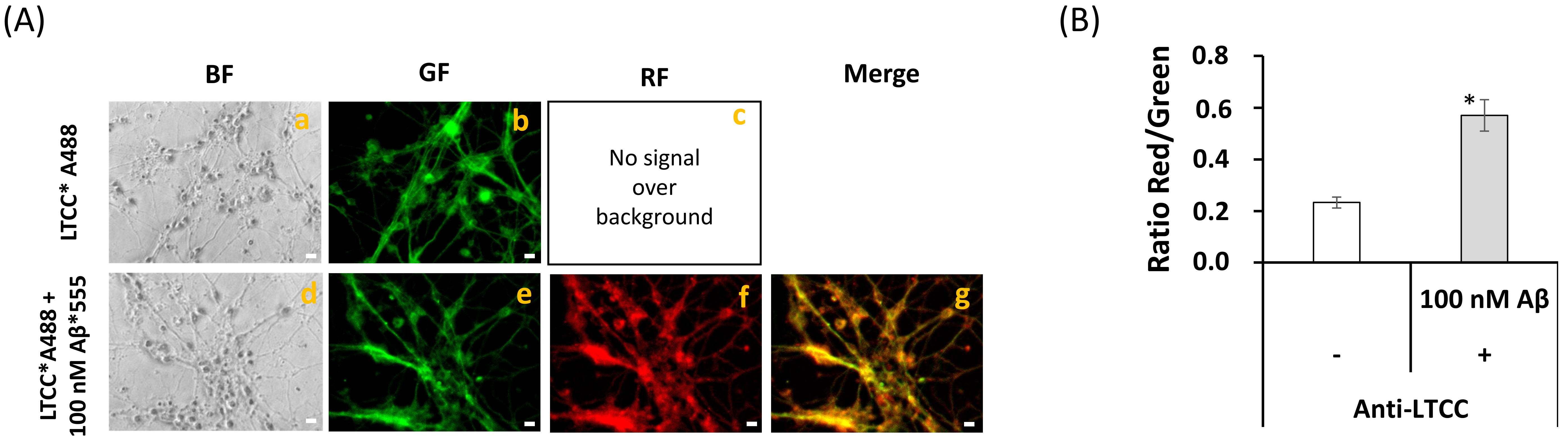
2.4. A Short Time Incubation with Submicromolar Concentrations of Aβ(1–42) Decreases the Resting Cytosolic Calcium Concentration through Inhibition of L-Type Calcium Channels (LTCCs) in Mature CGN
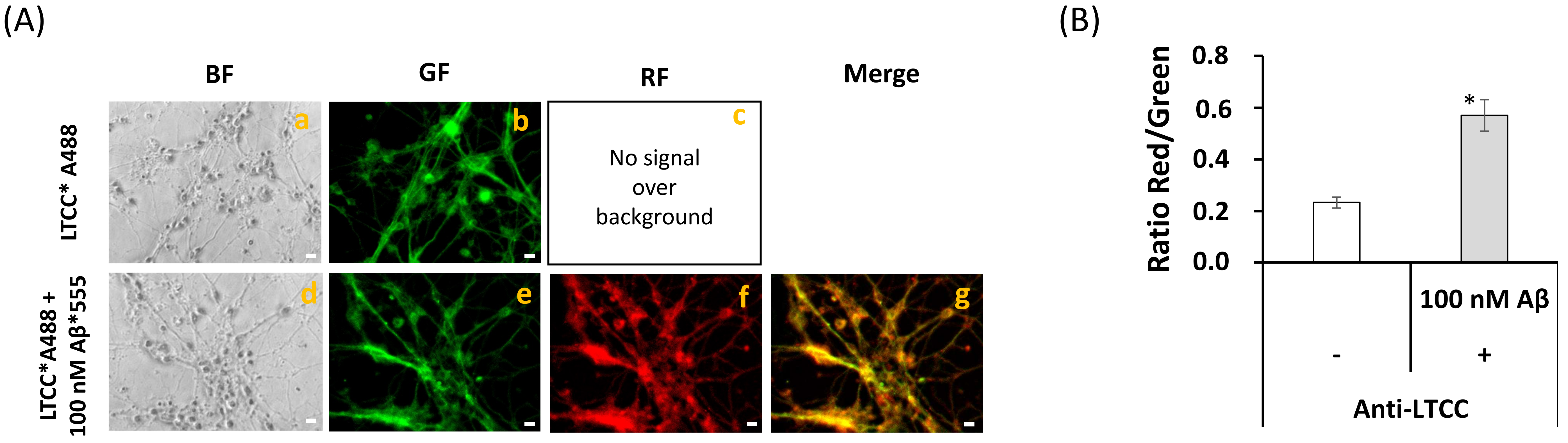
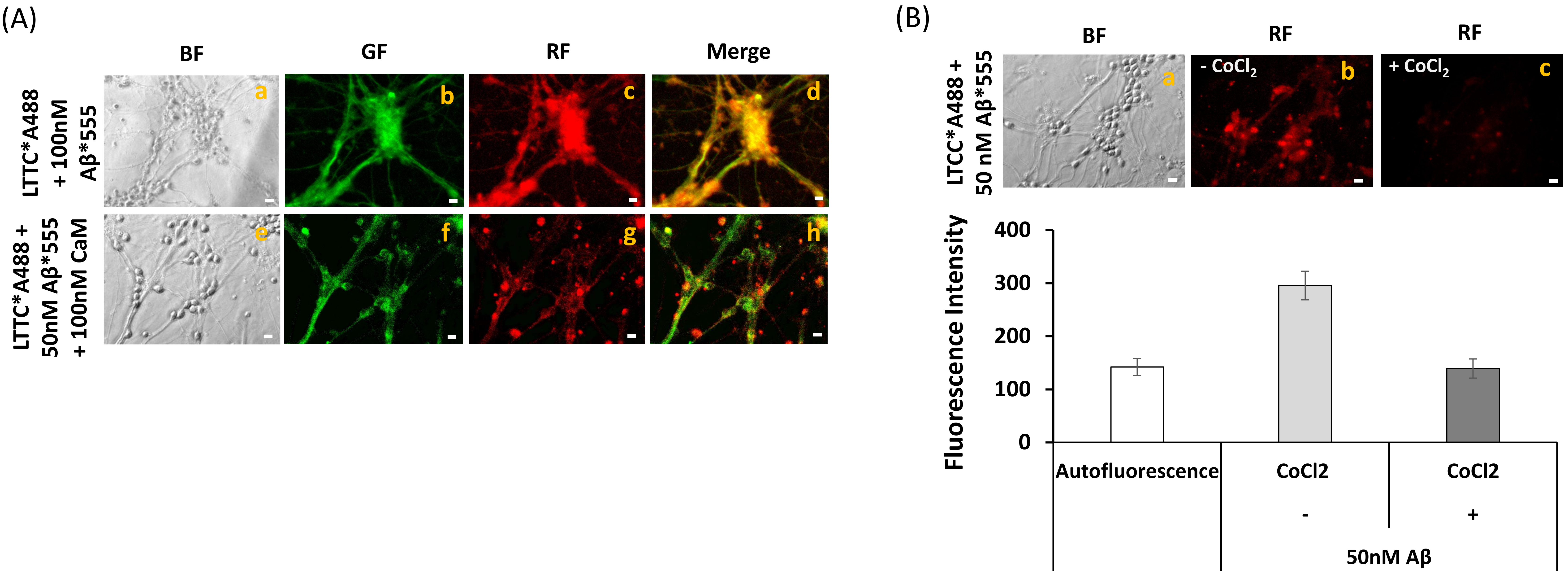
2.5. Extensive FRET between LTCCs Labeled with Alexa488 Fluorescent Antibody and Aβ(1–42)-HiLyteTM-Fluor555 in Fixed Mature CGN Is Largely Reversed by Addition of Exogenous CaM
3. Discussion
4. Materials and Methods
4.1. Preparation of Rat Cerebellar Granule Neurons (CGN)
4.2. Aβ(1–42) Solutions and Aggregation State
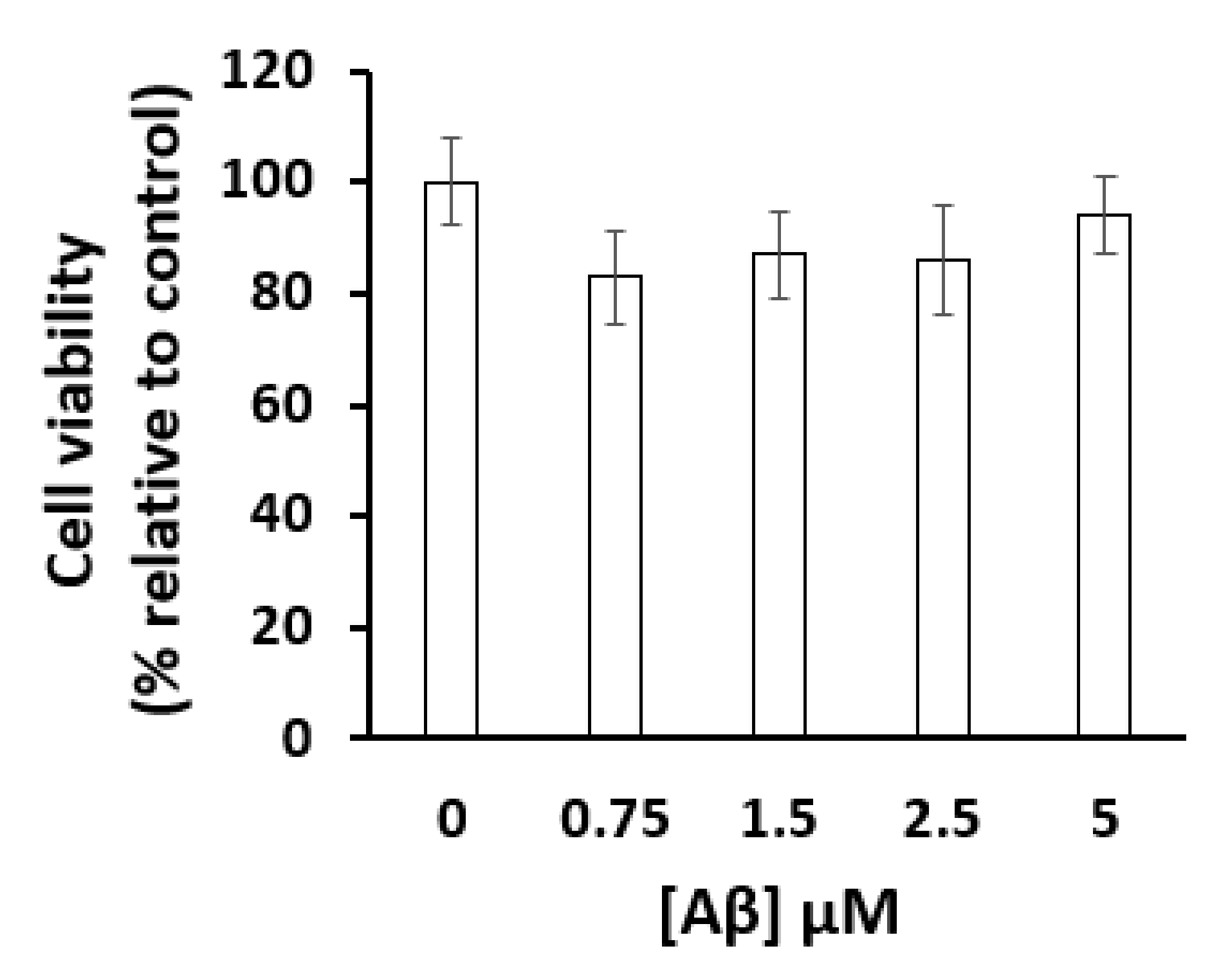
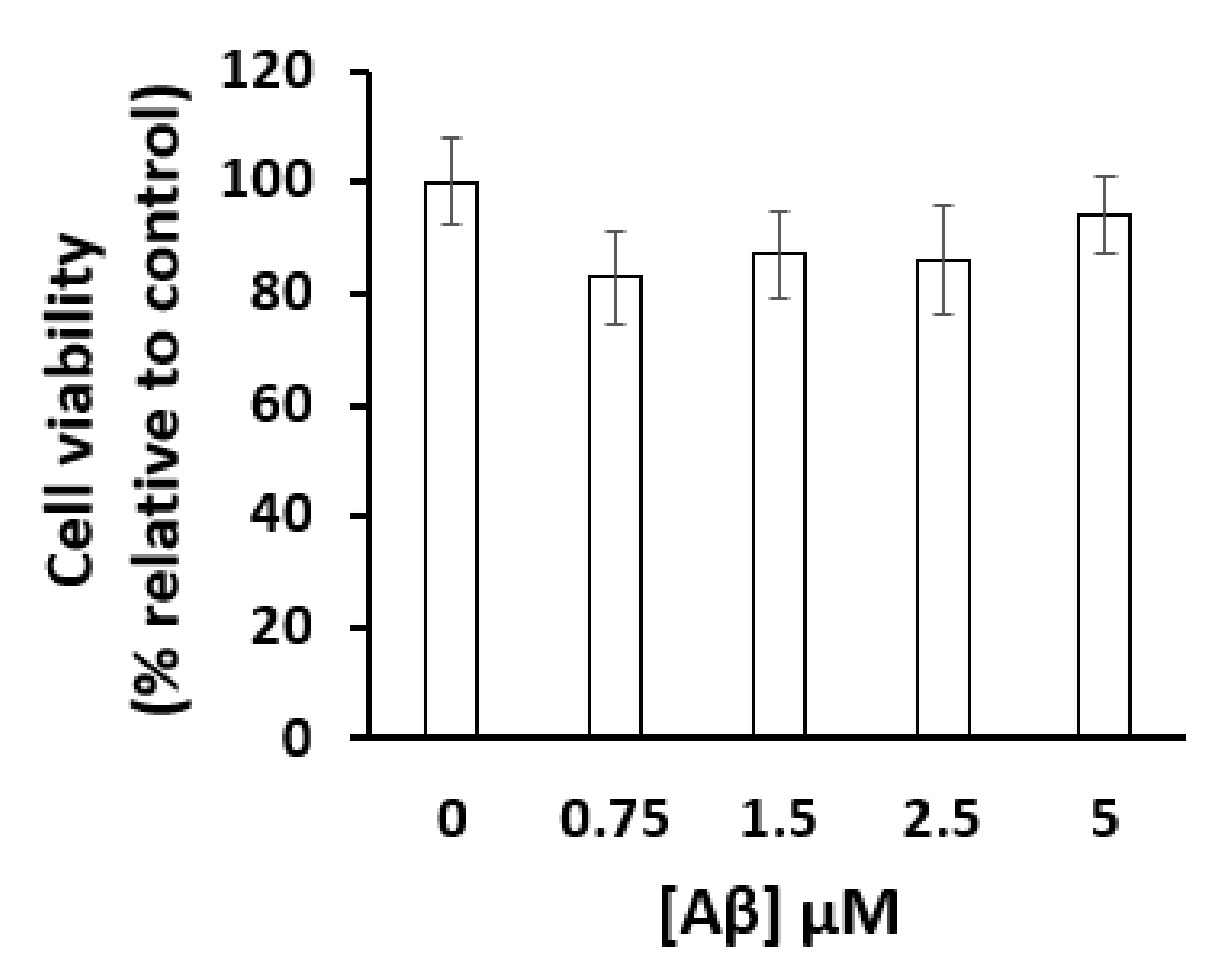
4.3. Cell Viability
4.4. Co-Immunoprecipitation
4.5. Western Blotting
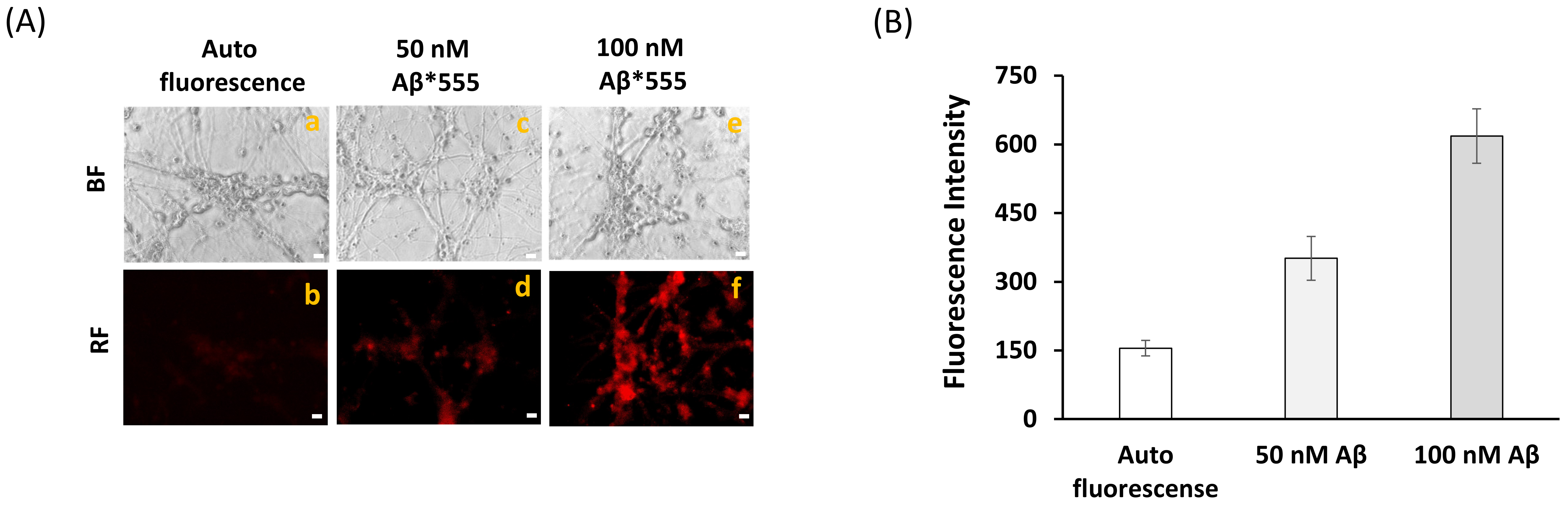
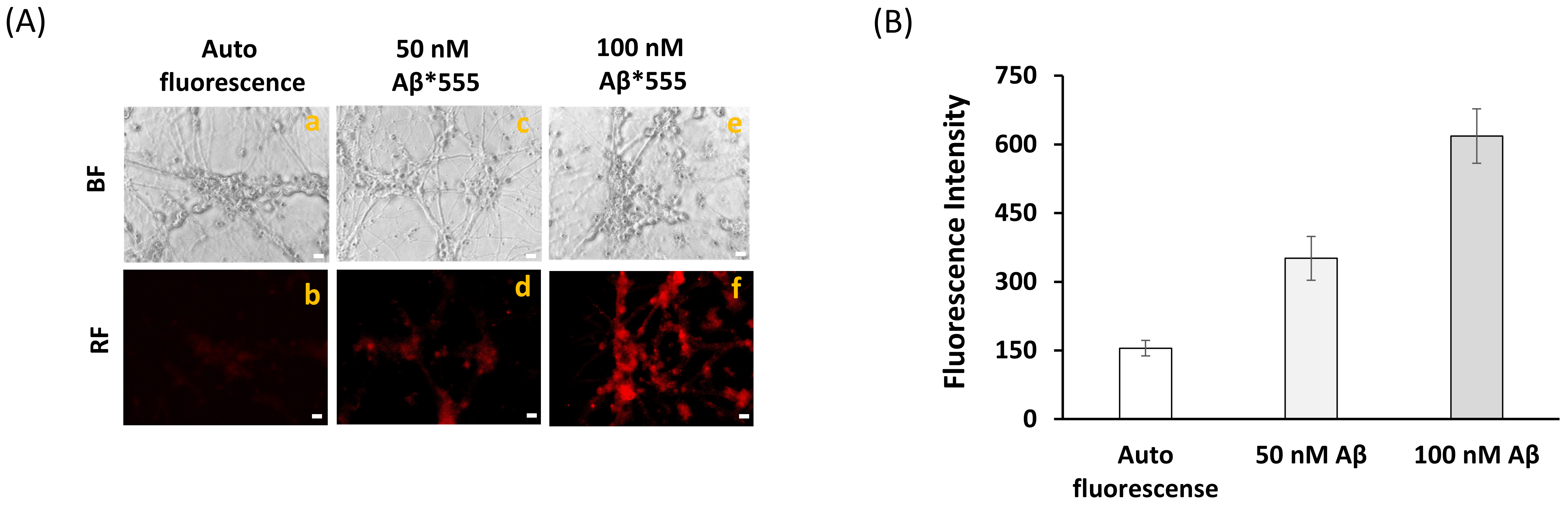
4.6. Measurements of Internalization of Aβ(1–42)-HiLyteTM-Fluor555 in Mature CGN
4.7. Fluorescence Resonance Energy Transfer (FRET) Imaging
4.8. Measurement of the Intracellular Free Ca2+ Concentration ([Ca2+]i)
4.9. Chemicals and Reagents
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid β peptide |
| AD | Alzheimer’s disease |
| Anti-CaM*A488/Anti-Cav1*A488/Anti-HRas*A488/Anti-LTCC*A488/Anti-PrPc*A488 | primary antibody of target protein (CaM, Cav1, HRas, LTCC or PrPc) stained with a secondary Alexa488 fluorescent antibody |
| Anti-HRas*Cy3 | primary antibody of HRas stained with a secondary Cy3 fluorescent antibody |
| BSA | bovine serum albumin |
| [Ca2+]i | intracellular free calcium concentration |
| CaM | calmodulin |
| CaMKII | Ca2+/calmodulin-dependent protein kinase isoform II |
| CaMBDs | calmodulin binding domains |
| CGN | cerebellar granule neurons |
| DMEM | Dulbecco’s Modified Eagle medium |
| FRET | fluorescence resonance energy transfer |
| Fura2-AM | Fura2 acetoxymethyl ester |
| LTCC | L-type calcium channels |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra¬zolium bromide |
| nNOS | neuronal isoform of nitric oxide synthase |
| PAGE | polyacrylamide gel electrophoresis |
| PBS | phosphate-buffered saline |
| PBST | PBS supplemented with 0.2% Triton X-100 |
| PMCA | plasma membrane calcium pump |
| PrPc | cellular prion protein |
| PVDF | polyvinylidene difluoride |
| ROI | region of interest |
| SDS | sodium dodecyl sulfate |
| TBS | Tris-buffered saline |
| TBST | TBS supplemented with 0.05% polyoxyethylene sorbitan monolaurate (Tween 20) |
| Tris | tris-(hydroxymethyl) aminomethane |
| Triton X-100 | 4-(1,1,3,3-tetramethyl butyl)phenyl-poly-ethylene glycol |
References
- Pike, L.J. Rafts defined: A report on the Keystone symposium on lipid rafts and cell function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Choo-Smith, L.-P.; Surewicz, W.K. The interaction between Alzheimer amyloid β(1–40) peptide and ganglioside GM1-containing membranes. FEBS Lett. 1997, 402, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Choo-Smith, L.-P.; Garzon-Rodriguez, W.; Glabe, C.G.; Surewicz, W.K. Acceleration of Amyloid Fibril Formation by Specific Binding of Aβ-(1–40) Peptide to Ganglioside-containing Membrane Vesicles. J. Biol. Chem. 1997, 272, 22987–22990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariga, T.; Kobayashi, K.; Hasegawa, A.; Kiso, M.; Ishida, H.; Miyatake, T. Characterization of high-affinity binding between gangliosides and amyloid β-protein. Arch. Biochem. Biophys. 2001, 388, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Aβ polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 868–877. [Google Scholar] [CrossRef]
- Kakio, A.; Nishimoto, S.I.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Cholesterol dependent formation of GM1 gangli-oside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 2001, 276, 24985–24990. [Google Scholar] [CrossRef] [Green Version]
- Kakio, A.; Nishimoto, S.I.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: Importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 2002, 41, 7385–7390. [Google Scholar] [CrossRef]
- Wood, W.; Schroeder, F.; Igbavboa, U.; Avdulov, N.; Chochina, S.V.; Avdulov, N. Brain membrane cholesterol domains, aging and amyloid beta-peptides. Neurobiol. Aging 2002, 23, 685–694. [Google Scholar] [CrossRef]
- Williamson, R.; Usardi, A.; Hanger, D.P.; Anderton, B.H. Membrane-bound β-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J. 2008, 22, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Lai, A.Y.; McLaurin, J. Mechanisms of Amyloid-Beta Peptide Uptake by Neurons: The Role of Lipid Rafts and Lipid Raft-Associated Proteins. Int. J. Alzheimer’s Dis. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younkin, S.G. The role of Aβ42 in Alzheimer’s disease. J. Physiol. Paris 1998, 92, 289–292. [Google Scholar] [CrossRef]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.-M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-β peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Kedia, N.; Illes-Toth, E.; Haralampiev, I.; Prisner, S.; Herrmann, A.; Wanker, E.E.; Bieschke, J. Amyloid-β(1–42) Ag-gregation Initiates Its Cellular Uptake and Cytotoxicity. J. Biol. Chem. 2016, 291, 19590–19606. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nat. Cell Biol. 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; la Ferla, F.M. Intraneuronal Aβ Causes the Onset of Early Alzheimer’s Disease-Related Cognitive Deficits in Transgenic Mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [Green Version]
- La Ferla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLachlan, D.R.C.; Wong, L.; Bergeron, C.; Baimbridge, K.G. Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1987, 1, 171–179. [Google Scholar] [CrossRef]
- Corbacho, I.; Berrocal, M.; Török, K.; Mata, A.M.; Gutierrez-Merino, C. High affinity binding of amyloid β-peptide to cal-modulin: Structural and functional implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef] [Green Version]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2013, 592, 281–293. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef] [Green Version]
- De Muro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-da-Silva, D.; Gutierrez-Merino, C. Caveolin-rich lipid rafts of the plasma membrane of mature cerebellar granule neurons are microcompartments for calcium/reactive oxygen and nitrogen species cross-talk signaling. Cell Calcium 2014, 56, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Merino, C.; Marques-da-Silva, D.; Fortalezas, S.; Samhan-Arias, A.K. Cytosolic calcium homeostasis in neurons: Control systems, modulation by reactive oxygen and nitrogen species, and space and time fluctuations. In Neurochemistry; Heinbockel, T., Ed.; InTech: Rijeka, Croatia, 2014; Chapter 3; pp. 59–110. [Google Scholar]
- Gutiérrez-Martín, Y.; Martín-Romero, F.J.; Henao, F.; Gutiérrez-Merino, C. Alteration of cytosolic free calcium homeostasis by SIN-1: High sensitivity of L-type Ca2+ channels to extracellular oxidative/nitrosative stress in cerebellar granule cells. J. Neurochem. 2005, 92, 973–989. [Google Scholar] [CrossRef]
- Gutierrez-Merino, C.; Marques-Da-Silva, D.; Fortalezas, S.; Samhan-Arias, A.K. The critical role of lipid rafts nanodomains in the cross-talk between calcium and reactive oxygen and nitrogen species in cerebellar granule neurons apoptosis by extracel-lular potassium deprivation. AIMS Mol. Sci. 2016, 3, 12–29. [Google Scholar] [CrossRef]
- Fortalezas, S.; Marques-da-Silva, D.; Gutierrez-Merino, C. Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons. Int. J. Mol. Sci. 2018, 19, 3667. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.Z.; de Maria, C.D.; Yue, D.T. Calmodulin Is the Ca2+ Sensor for Ca2+-Dependent Inactivation of L-Type Calcium Channels. Neuron 1999, 22, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Zühlke, R.D.; Pitt, G.S.; Deisseroth, K.; Tsien, R.W.; Reuter, H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nat. Cell Biol. 1999, 399, 159–162. [Google Scholar] [CrossRef]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E.; Calì, T. The plasma membrane calcium pumps: Focus on the role in (neuro)pathology. Biochem. Biophys. Res. Commun. 2017, 483, 1116–1124. [Google Scholar] [CrossRef]
- Mata, A.M. Functional interplay between plasma membrane Ca 2+ -ATPase, amyloid β-peptide and tau. Neurosci. Lett. 2018, 663, 55–59. [Google Scholar] [CrossRef]
- Suzuki, T.; Du, F.; Tian, Q.-B.; Zhang, J.; Endo, S. Ca2+/calmodulin-dependent protein kinase IIα clusters are associated with stable lipid rafts and their formation traps PSD-95. J. Neurochem. 2007, 104, 596–610. [Google Scholar] [CrossRef]
- Sato, Y.; Sagami, I.; Shimizu, T. Identification of Caveolin-1-interacting Sites in Neuronal Nitric-oxide Synthase. J. Biol. Chem. 2004, 279, 8827–8836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhu, D.-Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Marques-da-Silva, D.; Gutierrez-Merino, C. L-type voltage-operated calcium channels, N-methyl-d-aspartate receptors and neuronal nitric-oxide synthase form a calcium/redox nano-transducer within lipid rafts. Biochem. Biophys. Res. Commun. 2012, 420, 257–262. [Google Scholar] [CrossRef]
- Zhao, D.; Watson, J.B.; Xie, C.-W. Amyloid β Prevents Activation of Calcium/Calmodulin-Dependent Protein Kinase II and AMPA Receptor Phosphorylation During Hippocampal Long-Term Potentiation. J. Neurophysiol. 2004, 92, 2853–2858. [Google Scholar] [CrossRef]
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Marcos, D.; Sepúlveda, M.R.; Pérez, M.; Ávila, J.; Mata, A.M. Altered Ca 2+ dependence of synaptosomal plasma membrane Ca 2+ -ATPase in human brain affected by Alzheimer’s disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Padayachee, E.; Whiteley, C.G. Spectrofluorimetric analysis of the interaction of amyloid peptides with neuronal nitric oxide synthase: Implications in Alzheimer’s disease. Biochim. Biophys. Acta 2011, 1810, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Padayachee, E.; Ngqwala, N.; Whiteley, C.G. Association of β-amyloid peptide fragments with neuronal nitric oxide synthase: Implications in the etiology of Alzheimers disease. J. Enzym. Inhib. Med. Chem. 2011, 27, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Wankerl, K.; Weise, D.; Gentner, R.; Rumpf, J.-J.; Classen, J. L-Type Voltage-Gated Ca2+ Channels: A Single Molecular Switch for Long-Term Potentiation/Long-Term Depression-Like Plasticity and Activity-Dependent Metaplasticity in Humans. J. Neurosci. 2010, 30, 6197–6204. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.M.; Bartsch, D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014, 357, 463–476. [Google Scholar] [CrossRef]
- O’Day, D.H.; Myre, M.A. Calmodulin-binding domains in Alzheimer’s disease proteins: Extending the calcium hypothesis. Biochem. Biophys. Res. Commun. 2004, 320, 1051–1054. [Google Scholar] [CrossRef]
- Sepúlveda, M.R.; Berrocal-Carrillo, M.; Gasset, M.; Mata, A.M. The Plasma Membrane Ca2+-ATPase Isoform 4 Is Localized in Lipid Rafts of Cerebellum Synaptic Plasma Membranes. J. Biol. Chem. 2006, 281, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Marques-Da-Silva, D.; Samhan-Arias, A.K.; Tiago, T.; Gutiérrez-Merino, C. L-type calcium channels and cytochrome b5 reductase are components of protein complexes tightly associated with lipid rafts microdomains of the neuronal plasma membrane. J. Proteom. 2010, 73, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Garcia-Bereguiain, M.A.; Martin-Romero, F.J.; Gutierrez-Merino, C. Clustering of plasma membrane-bound cytochrome b5 reductase within ’lipid rafts’ microdomains of the neuronal plasma membrane. Mol. Cell. Neurosci. 2009, 40, 14–26. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Marques-da-Silva, D.; Yanamala, N.; Gutierrez-Merino, C. Stimulation and clustering of cytochrome b5 reductase in caveolin-rich lipid microdomains is an early event in oxidative stress-mediated apoptosis of cerebellar granule neurons. J. Proteom. 2012, 75, 2934–2949. [Google Scholar] [CrossRef]
- Cuenda, A.; Henao, F.; Gutierrez-Merino, C. Distances between functional sites of the Ca2+ + Mg2+-ATPase from sarcoplasmic reticulum using Co2+ as a spectroscopic ruler. JBIC J. Biol. Inorg. Chem. 1990, 194, 663–670. [Google Scholar] [CrossRef]
- Nguyen, D.M.; Chen, L.S.; Jeng, G.; Yu, W.-P.; Chen, T.-Y. Cobalt ion interaction with TMEM16A calcium-activated chloride channel: Inhibition and potentiation. PLoS ONE 2020, 15, e0231812. [Google Scholar] [CrossRef] [Green Version]
- Highsmith, S.; Murphy, A.J. Nd3+ and Co2+ binding to sarcoplasmic reticulum CaATPase. An estimation of the distance from the ATP binding site to the high-affinity calcium binding sites. J. Biol. Chem. 1984, 259, 14651–14656. [Google Scholar] [CrossRef]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between Human Prion Protein and Amyloid-(Aβ) Oligomers. Role of n-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhao, Y.; Zhang, L.; Yu, W.; Wang, Y.; Chang, W. Cellular Prion Protein as a Receptor of Toxic Amyloid-β42 Oligomers Is Important for Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudmon, A.; Schulman, H.; Kim, J.; Maltez, J.M.; Tsien, R.W.; Pitt, G.S. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 2005, 171, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.S.; Karl, R.; Moosmang, S.; Lenhardt, P.; Klugbauer, N.; Hofmann, F.; Kleppisch, T.; Welling, A. Calmodulin kinase II is involved in voltage dependent facilitation of the L-type Cav1.2 calcium channel: Identification of the phosphorylation sites. J. Biol. Chem. 2006, 281, 25560–25567. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.Y. Calmodulin. Sci. Am. 1982, 246, 62–70. [Google Scholar] [CrossRef]
- Biber, A.; Schmid, G.; Hempel, K. Calmodulin content in specific brain areas. Exp. Brain Res. 1984, 56, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nat. Cell Biol. 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, C.J.; McGauran, G.; Willén, K.; Gouras, G.K.; O’Connell, D.J.; Linse, S. Direct High Affinity Interaction between Aβ42 and GSK3α Stimulates Hyperphosphorylation of Tau. A New Molecular Link in Alzheimer’s Disease? ACS Chem. Neurosci. 2015, 7, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, C.; Nichino, D.; Zampagni, M.; Bernacchioni, C.; Evangelisti, E.; Pensalfini, A.; Liguri, G.; Gliozzi, A.; Stefani, M.; Relini, A. A protective role for lipid raft cholesterol against amyloid-induced membrane damage in human neuroblastoma cells. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2204–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Sepulveda, M.R.; Vazquez-Hernandez, M.; Mata, A.M. Calmodulin antagonizes amyloid-β peptides-mediated inhibition of brain plasma membrane Ca2+-ATPase. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 961–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobi-lization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emilsson, L.; Saetre, P.; Jazin, E. Alzheimer’s disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol. Dis. 2006, 21, 618–625. [Google Scholar] [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.-F.; Hao, Y.-H.; Serneels, L.; de Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Sun, S.; Herreman, A.; de Strooper, B.; Bezprozvanny, I. Role of Presenilins in Neuronal Calcium Homeostasis. J. Neurosci. 2010, 30, 8566–8580. [Google Scholar] [CrossRef] [Green Version]
- Fortalezas, S.; Marques-Da-Silva, D.; Gutierrez-Merino, C. Creatine protects against cytosolic calcium dysregulation, mitochondrial depolarization and increase of reactive oxygen species production in rotenone-induced cell death of cerebellar granule neurons. Neurotox. Res. 2018, 34, 717–732. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Martin-Romero, F.J.; Gutierrez-Merino, C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for the plasma membrane NADH oxidase activity in the commitment of apoptosis. Free Radic. Biol. Med. 2004, 37, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Fancy, D.A.; Kodadek, T. Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl. Acad. Sci. USA 1999, 96, 6020–6024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitan, G.; Teplow, D.B. Rapid Photochemical Cross-Linking: A New Tool for Studies of Metastable, Amyloidogenic Protein Assemblies. Acc. Chem. Res. 2004, 37, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; von Jagow, G. Tricine–sodium dodecyl sulfate polyacrylamide gel electrophoresis for the separation of proteins in the range from 1–100 kDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef]
- Saavedra, L.; Mohamed, A.; Ma, V.; Kar, S.; de Chaves, E.P. Internalization of β-Amyloid Peptide by Primary Neurons in the Absence of Apolipoprotein E. J. Biol. Chem. 2007, 282, 35722–35732. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. Binding of Amyloid β(1–42)-Calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis. Int. J. Mol. Sci. 2021, 22, 1984. https://doi.org/10.3390/ijms22041984
Poejo J, Salazar J, Mata AM, Gutierrez-Merino C. Binding of Amyloid β(1–42)-Calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis. International Journal of Molecular Sciences. 2021; 22(4):1984. https://doi.org/10.3390/ijms22041984
Chicago/Turabian StylePoejo, Joana, Jairo Salazar, Ana M. Mata, and Carlos Gutierrez-Merino. 2021. "Binding of Amyloid β(1–42)-Calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis" International Journal of Molecular Sciences 22, no. 4: 1984. https://doi.org/10.3390/ijms22041984
APA StylePoejo, J., Salazar, J., Mata, A. M., & Gutierrez-Merino, C. (2021). Binding of Amyloid β(1–42)-Calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis. International Journal of Molecular Sciences, 22(4), 1984. https://doi.org/10.3390/ijms22041984