
Costunolide, a Sesquiterpene Lactone, Suppresses Skin Cancer via Induction of Apoptosis and Blockage of Cell Proliferation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
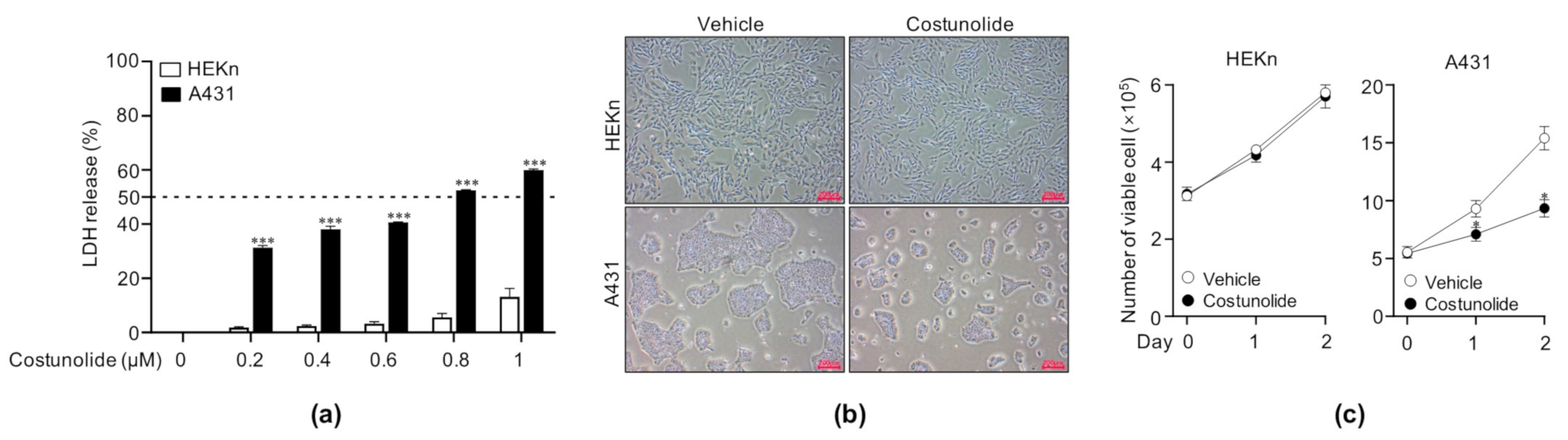
2.1. Costunolide Reduced Cell Viability in A431 Cells
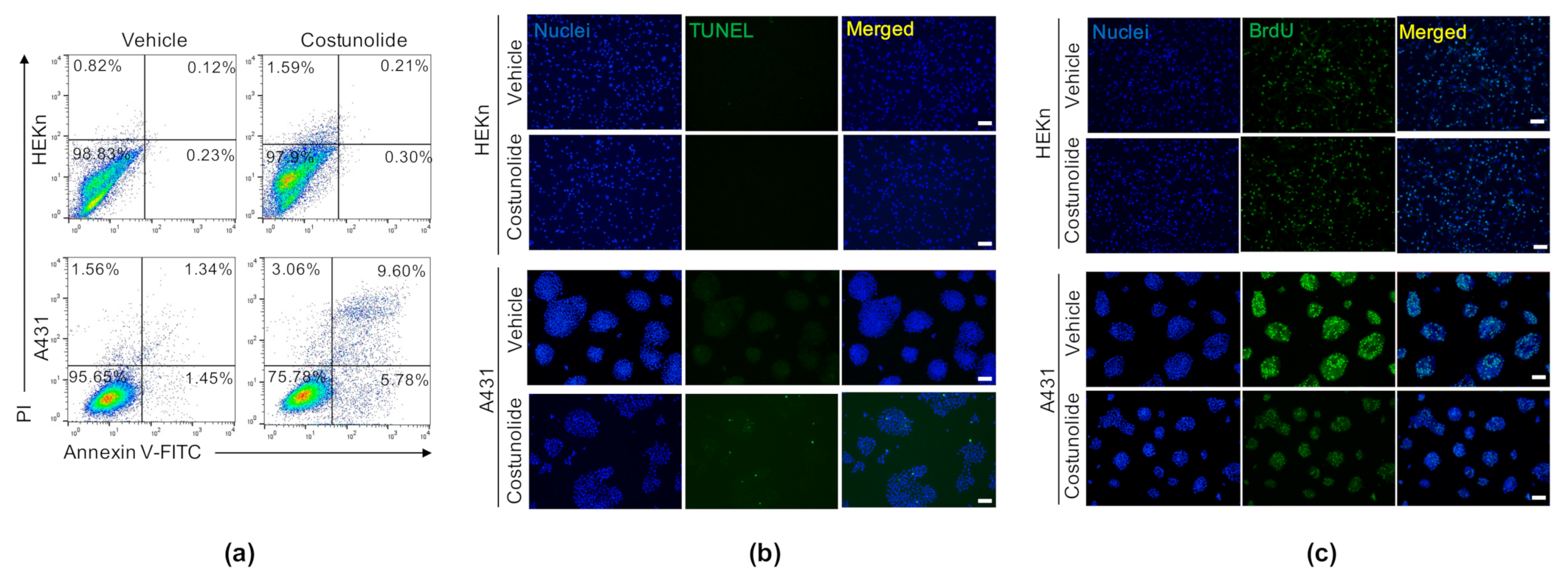
2.2. Costunolide Induced Apoptosis and Suppressed Proliferation in A431 Cells
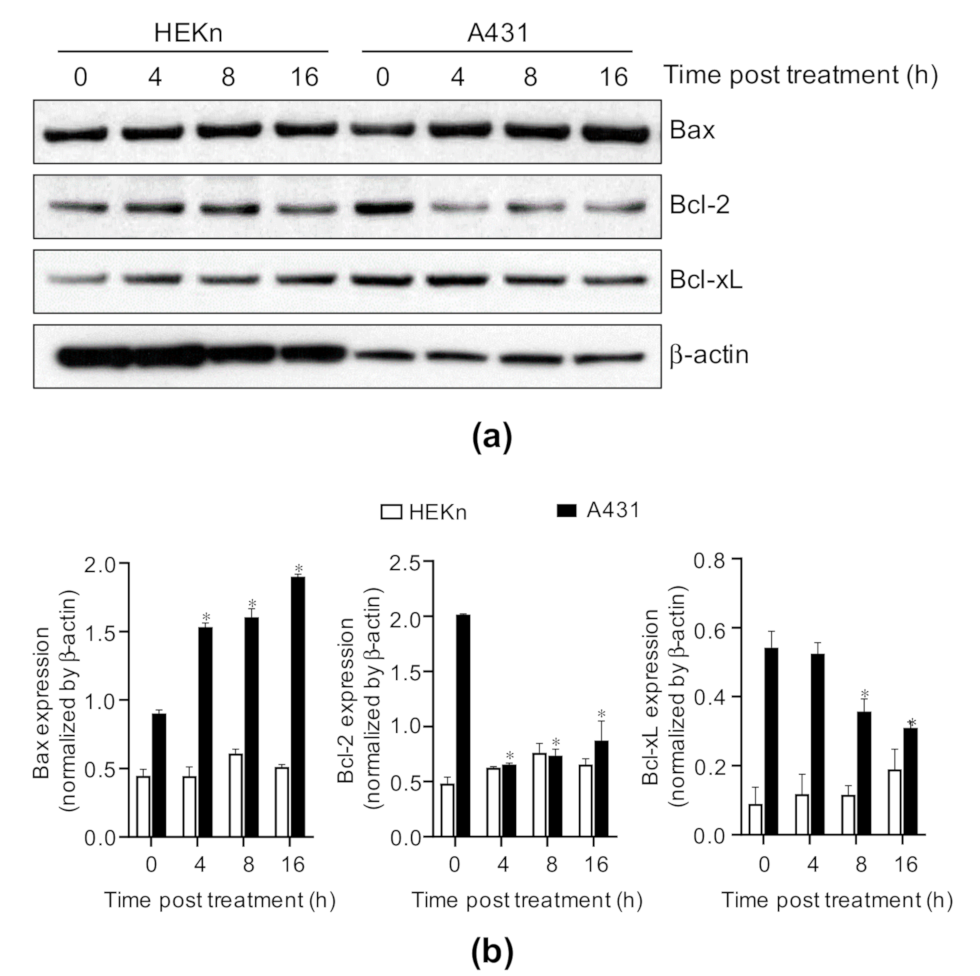
2.3. Costunolide Regulates the Bcl-2 Family Proteins in A431 Cells
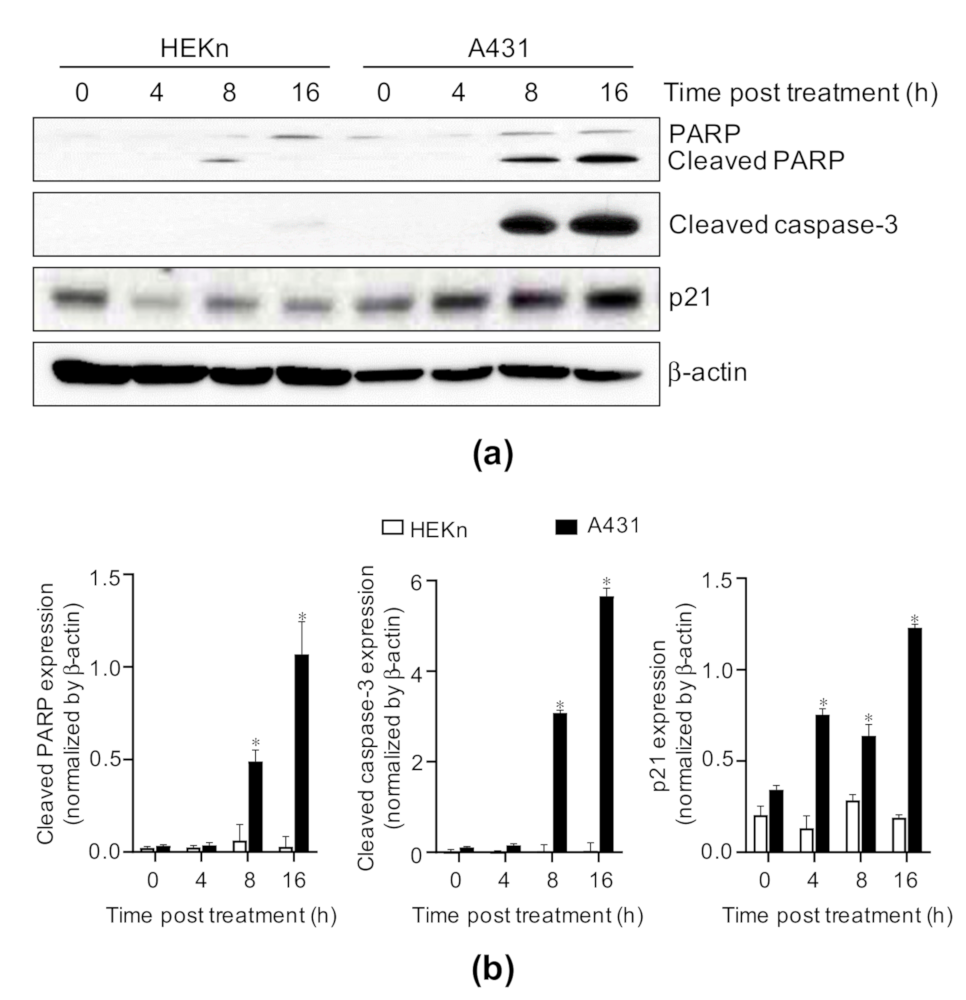
2.4. Costunolide Triggered Caspase-3 Activation, Poly-ADP Ribose Polymerase (PARP) Cleavage, and Increase in p21 Expression in A431 Cells
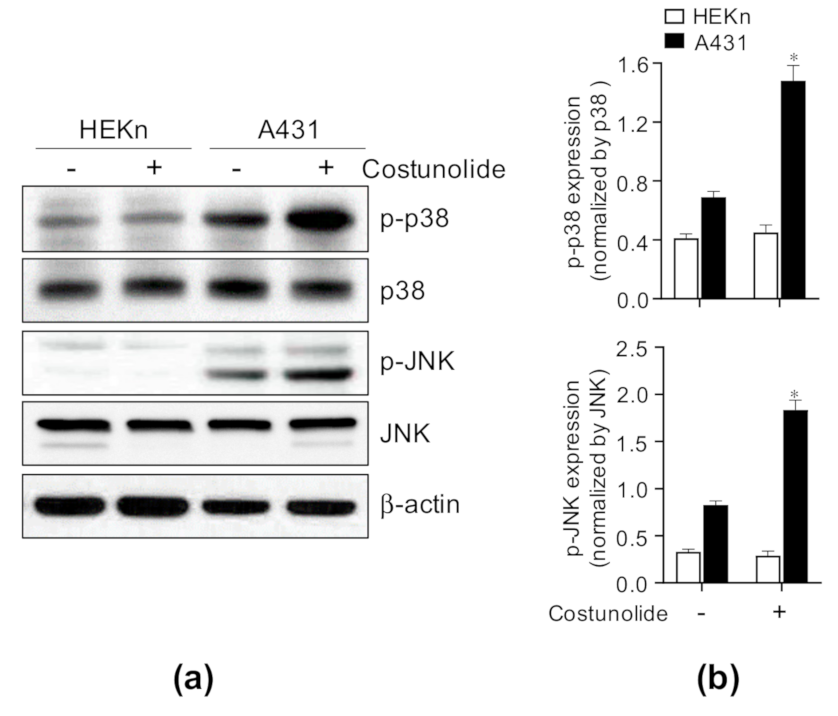
2.5. Costunolide Triggered Apoptosis via Activation of JNK and p38 Signaling in A431 Cells
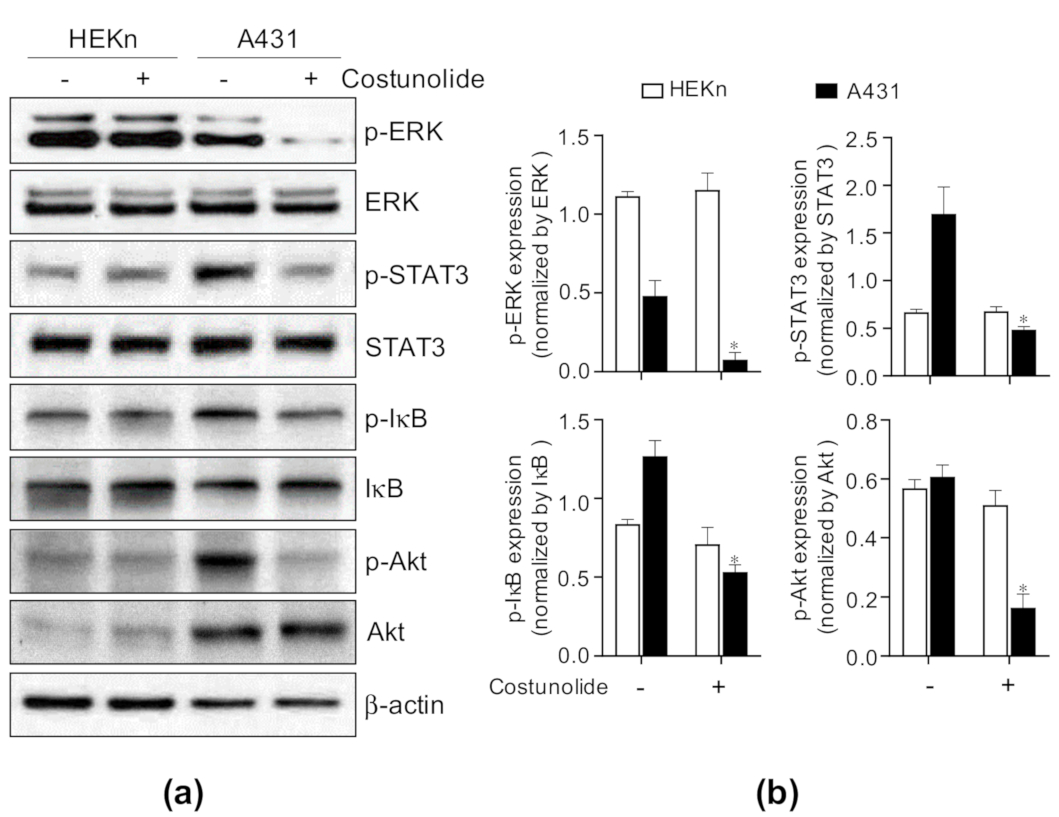
2.6. Costunolide Suppressed Cell Proliferation and Survival via Inhibiting ERK, STAT3, NF-κB, and Akt Activation in A431 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cytotoxicity Assay
control)]/[(OD450positive control − OD450background) − (OD450negative control − OD450background control)] × 100
4.4. Trypan Blue Exclusion Assay
4.5. Analysis of Apoptotic Cell Death
4.6. TUNEL Assay
4.7. BrdU Incorporation Assay
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fahradyan, A.; Howell, A.C.; Wolfswinkel, E.M.; Tsuha, M.; Sheth, P.; Wong, A.K. Updates on the Management of Non-Melanoma Skin Cancer (NMSC). Healthcare 2017, 5, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzuka, A.G.; Book, S.E. Basal Cell Carcinoma: Pathogenesis, Epidemiology, Clinical Features, Diagnosis, Histopathology, and Management. Yale J. Biol. Med. 2015, 88, 167–179. [Google Scholar]
- Eisemann, N.; Waldmann, A.; Geller, A.C.; Weinstock, M.A.; Volkmer, B.; Greinert, R.; Breitbart, E.W.; Katalinic, A. Non-Melanoma Skin Cancer Incidence and Impact of Skin Cancer Screening on Incidence. J. Investig. Dermatol. 2014, 134, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Tarallo, M.; Cigna, E.; Frati, R.; Delfino, S.; Innocenzi, D.; Fama, U.; Corbianco, A.; Scuderi, N. Metatypical Basal Cell Carcinoma: A Clinical Review. J. Exp. Clin. Cancer Res. 2008, 27, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jennings, L.; Schmults, C.D. Management of High-Risk Cutaneous Squamous Cell Carcinoma. J. Clin. Aesthet. Dermatol. 2010, 3, 39–48. [Google Scholar] [PubMed]
- Gupta, A.K.; Rather, M.A.; Kumar Jha, A.; Shashank, A.; Singhal, S.; Sharma, M.; Pathak, U.; Sharma, D.; Mastinu, A. Artocarpus Lakoocha Roxb. and Artocarpus Heterophyllus Lam. Flowers: New Sources of Bioactive Compounds. Plants 2020, 9, 1329. [Google Scholar] [CrossRef] [PubMed]
- Mastinu, A.; Bonini, S.A.; Rungratanawanich, W.; Aria, F.; Marziano, M.; Maccarinelli, G.; Abate, G.; Premoli, M.; Memo, M.; Uberti, D. Gamma-Oryzanol Prevents LPS-Induced Brain Inflammation and Cognitive Impairment in Adult Mice. Nutrients 2019, 11, 728. [Google Scholar] [CrossRef] [Green Version]
- Amina, M.; Al Musayeib, N.M.; Alarfaj, N.A.; El-Tohamy, M.F.; Oraby, H.F.; Al Hamoud, G.A.; Bukhari, S.I.; Moubayed, N.M.S. Biogenic Green Synthesis of MgO Nanoparticles Using Saussurea Costus Biomasses for a Comprehensive Detection of Their Antimicrobial, Cytotoxicity against MCF-7 Breast Cancer Cells and Photocatalysis Potentials. PLoS ONE 2020, 15, e0237567. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.-F.; Su, J.; Pan, Z.-H.; Zhang, Z.-J.; Li, X.-N.; Song, L.-D.; Wu, X.-D.; Zhao, Q.-S. Cytotoxic Sesquiterpenoids from the Leaves of Magnolia Grandiflora. Phytochemistry 2018, 155, 182–190. [Google Scholar] [CrossRef]
- Pacifico, S.; Gallicchio, M.; Lorenz, P.; Potenza, N.; Galasso, S.; Marciano, S.; Fiorentino, A.; Stintzing, F.C.; Monaco, P. Apolar Laurus Nobilis Leaf Extracts Induce Cytotoxicity and Apoptosis towards Three Nervous System Cell Lines. Food Chem. Toxicol. 2013, 62, 628–637. [Google Scholar] [CrossRef]
- Pandey, M.M.; Rastogi, S.; Rawat, A.K.S. Saussurea Costus: Botanical, Chemical and Pharmacological Review of an Ayurvedic Medicinal Plant. J. Ethnopharmacol. 2007, 110, 379–390. [Google Scholar] [CrossRef]
- Li, A.; Sun, A.; Liu, R. Preparative Isolation and Purification of Costunolide and Dehydrocostuslactone from Aucklandia Lappa Decne by High-Speed Counter-Current Chromatography. J. Chromatogr. A 2005, 1076, 193–197. [Google Scholar] [CrossRef]
- De Marino, S.; Borbone, N.; Zollo, F.; Ianaro, A.; Di Meglio, P.; Iorizzi, M. New Sesquiterpene Lactones from Laurus nobilis Leaves as Inhibitors of Nitric Oxide Production. Planta Med. 2005, 71, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.H.; Lee, J.-H.; Park, Y.J.; Hong, Y.-S.; Kim, H.S.; Kim, K.-W.; Lee, J.J. A Sesquiterpene Lactone, Costunolide, from Magnolia Grandiflora Inhibits NF-ΚB by Targeting IκB Phosphorylation. Planta Med. 2001, 67, 103–107. [Google Scholar] [CrossRef]
- Mondranondra, I.; Che, C.; Rimando, A.M.; Vajrodaya, S.; Fong, H.H.S.; Farnsworth, N.R. Sesquiterpene Lactones and Other Constituents from a Cytotoxic Extract of Michelia Floribunda. Pharm. Res. 1990, 7, 1269–1272. [Google Scholar] [CrossRef]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/Endoplasmic Reticulum Calcium ATPase Inhibitors: Beyond Anticancer Perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Drašar, P.; Rimpelová, S.; Christensen, S.B.; Khripach, V.A.; Jurášek, M. Large Scale Conversion of Trilobolide into the Payload of Mipsagargin: 8-O-(12-Aminododecanoyl)-8-O-Debutanoylthapsigargin. Biomolecules 2020, 10, 1640. [Google Scholar] [CrossRef]
- Jurášek, M.; Rimpelová, S.; Kmoníčková, E.; Drašar, P.; Ruml, T. Tailor-Made Fluorescent Trilobolide to Study Its Biological Relevance. J. Med. Chem. 2014, 57, 7947–7954. [Google Scholar] [CrossRef]
- Eliza, J.; Daisy, P.; Ignacimuthu, S. Antioxidant Activity of Costunolide and Eremanthin Isolated from Costus Speciosus (Koen Ex. Retz) Sm. Chem. Biol. Interact. 2010, 188, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Di Paola, R.; Suzuki, H.; Paterniti, I.; Ahmad, A.; Mariotto, S.; Cuzzocrea, S. Costunolide and Dehydrocostuslactone, Two Natural Sesquiterpene Lactones, Ameliorate the Inflammatory Process Associated to Experimental Pleurisy in Mice. Eur. J. Pharmacol. 2014, 730, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.-S.; Lim, H.-S.; Jeong, S.-J.; Shin, H.-K. Anti-Allergic Effects of Sesquiterpene Lactones from the Root of Aucklandia Lappa Decne. Mol. Med. Rep. 2015, 12, 7789–7795. [Google Scholar] [CrossRef] [Green Version]
- Ham, A.; Lee, S.-J.; Shin, J.; Kim, K.-H.; Mar, W. Regulatory Effects of Costunolide on Dopamine Metabolism-Associated Genes Inhibit Dopamine-Induced Apoptosis in Human Dopaminergic SH-SY5Y Cells. Neurosci. Lett. 2012, 507, 101–105. [Google Scholar] [CrossRef]
- Duraipandiyan, V.; Al-Harbi, N.A.; Ignacimuthu, S.; Muthukumar, C. Antimicrobial Activity of Sesquiterpene Lactones Isolated from Traditional Medicinal Plant, Costus Speciosus (Koen Ex.Retz.) Sm. BMC Complement. Altern. Med. 2012, 12, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Liu, L.; Yao, W. Activation of P53 by Costunolide Blocks Glutaminolysis and Inhibits Proliferation in Human Colorectal Cancer Cells. Gene 2018, 678, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Choi, E.M. Costunolide Stimulates the Function of Osteoblastic MC3T3-E1 Cells. Int. Immunopharmacol. 2011, 11, 712–718. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, Y.; Fan, J.; Lin, X.; Liu, C.; Xu, Y.; Ji, W.; Yan, C.; Su, C. Costunolide and Dehydrocostuslactone Combination Treatment Inhibit Breast Cancer by Inducing Cell Cycle Arrest and Apoptosis through C-Myc/P53 and AKT/14-3-3 Pathway. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.-L.; Pan, S.-L.; Ho, Y.-F.; Hwang, T.-L.; Kung, F.-L.; Guh, J.-H. Costunolide Induces Apoptosis through Nuclear Calcium2+ Overload and DNA Damage Response in Human Prostate Cancer. J. Urol. 2011, 185, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, B.; Zou, Z.; Li, W.; Zhang, Y.; Xie, J.; Liu, C. Costunolide Enhances Doxorubicin-Induced Apoptosis in Prostate Cancer Cells via Activated Mitogen-Activated Protein Kinases and Generation of Reactive Oxygen Species. Oncotarget 2017, 8, 107701–107715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-Y.; Chang, H.-S.; Chen, I.-S.; Chen, C.-J.; Hsu, M.-L.; Fu, S.-L.; Chen, Y.-J. Costunolide Causes Mitotic Arrest and Enhances Radiosensitivity in Human Hepatocellular Carcinoma Cells. Radiat. Oncol. Lond. Engl. 2011, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hua, P.; Zhang, G.; Zhang, Y.; Sun, M.; Cui, R.; Li, X.; Li, B.; Zhang, X. Costunolide Induces G1/S Phase Arrest and Activates Mitochondrial-Mediated Apoptotic Pathways in SK-MES 1 Human Lung Squamous Carcinoma Cells. Oncol. Lett. 2016, 11, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; He, X.; Yang, C. Costunolide Promotes Imatinib-Induced Apoptosis in Chronic Myeloid Leukemia Cells via the Bcr/Abl-Stat5 Pathway. Phytother. Res. 2018, 32, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of Apoptosis by TUNEL Assay. Methods Mol. Biol. Clifton 2012, 887, 41–47. [Google Scholar] [CrossRef]
- Cecchini, M.J.; Amiri, M.; Dick, F.A. Analysis of Cell Cycle Position in Mammalian Cells. J. Vis. Exp. JoVE 2012. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Cytochrome c in the Apoptotic and Antioxidant Cascades. FEBS Lett. 1998, 423, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Stingl, L.; Morrison, C.; Jantsch, M.; Los, M.; Schulze-Osthoff, K.; Wagner, E.F. PARP Is Important for Genomic Stability but Dispensable in Apoptosis. Genes Dev. 1997, 11, 2347–2358. [Google Scholar] [CrossRef] [Green Version]
- Soldani, C.; Scovassi, A.I. Poly(ADP-Ribose) Polymerase-1 Cleavage during Apoptosis: An Update. Apoptosis Int. J. Program. Cell Death 2002, 7, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple Functions of P21 in Cell Cycle, Apoptosis and Transcriptional Regulation after DNA Damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Nagata, Y.; Todokoro, K. Requirement of Activation of JNK and P38 for Environmental Stress-Induced Erythroid Differentiation and Apoptosis and of Inhibition of ERK for Apoptosis. Blood 1999, 94, 853–863. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-P38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Peng, Z.; Su, C. Potential Anti-Cancer Activities and Mechanisms of Costunolide and Dehydrocostuslactone. Int. J. Mol. Sci. 2015, 16, 10888–10906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Choi, B.Y. Costunolide—A Bioactive Sesquiterpene Lactone with Diverse Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 2926. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Lee, K.-T. Costunolide-Induced Apoptosis in Human Leukemia Cells: Involvement of c-Jun N-Terminal Kinase Activation. Biol. Pharm. Bull. 2009, 32, 1803–1808. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Korsmeyer, S.J. Mechanisms of Cytochrome c Release by Proapoptotic BCL-2 Family Members. Biochem. Biophys. Res. Commun. 2003, 304, 437–444. [Google Scholar] [CrossRef]
- Choi, Y.K.; Seo, H.S.; Choi, H.S.; Choi, H.S.; Kim, S.R.; Shin, Y.C.; Ko, S.-G. Induction of Fas-Mediated Extrinsic Apoptosis, P21WAF1-Related G2/M Cell Cycle Arrest and ROS Generation by Costunolide in Estrogen Receptor-Negative Breast Cancer Cells, MDA-MB-231. Mol. Cell. Biochem. 2012, 363, 119–128. [Google Scholar] [CrossRef]
- Niculescu, A.B.; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of P21(Cip1/Waf1) at Both the G1/S and the G2/M Cell Cycle Transitions: PRb Is a Critical Determinant in Blocking DNA Replication and in Preventing Endoreduplication. Mol. Cell. Biol. 1998, 18, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Manikkam, R. Cytotoxic Impact of Costunolide Isolated from Costus Speciosus on Breast Cancer via Differential Regulation of Cell Cycle-An In-Vitro and In-Silic o Approach: Anticancer Potentials of Costunolide on Breast Cancer. Phytother. Res. 2015, 29, 1532–1539. [Google Scholar] [CrossRef]
- Cai, H.; Li, L.; Jiang, J.; Zhao, C.; Yang, C. Costunolide Enhances Sensitivity of K562/ADR Chronic Myeloid Leukemia Cells to Doxorubicin through PI3K/Akt Pathway. Phytother. Res. 2019, 33, 1683–1688. [Google Scholar] [CrossRef]
- Rawlings, J.S. The JAK/STAT Signaling Pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 Activation in Colon Cancer Is Associated with Enhanced Cell Proliferation and Tumor Growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.-H.; Qin, L.; Li, X. Role of STAT3 Signaling Pathway in Breast Cancer. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Pedranzini, L.; Leitch, A.; Bromberg, J. Stat3 Is Required for the Development of Skin Cancer. J. Clin. Investig. 2004, 114, 619–622. [Google Scholar] [CrossRef]
- Yin, W.; Cheepala, S.; Roberts, J.N.; Syson-Chan, K.; DiGiovanni, J.; Clifford, J.L. Active Stat3 Is Required for Survival of Human Squamous Cell Carcinoma Cells in Serum-Free Conditions. Mol. Cancer 2006, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Crowe, P.J.; Goldstein, D.; Yang, J.-L. STAT3 Inhibition, a Novel Approach to Enhancing Targeted Therapy in Human Cancers (Review). Int. J. Oncol. 2012, 41, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Meloche, S.; Pouysségur, J. The ERK1/2 Mitogen-Activated Protein Kinase Pathway as a Master Regulator of the G1- to S-Phase Transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Wu, D.; Hirao, A.; Lahti, J.M.; Liu, L.; Mazza, B.; Kidd, V.J.; Mak, T.W.; Ingram, A.J. ERK Activation Mediates Cell Cycle Arrest and Apoptosis after DNA Damage Independently of P53. J. Biol. Chem. 2002, 277, 12710–12717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Ouyang, G.; Bao, S. The Activation of Akt/PKB Signaling Pathway and Cell Survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, Death, and Autophagy in Cancer: NF-ΚB Turns up Everywhere. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, J.-P.; Liu, X.; Tang, J.-J.; Xiang, P.; Ma, X.-M. Semisynthesis, an Anti-Inflammatory Effect of Derivatives of 1β-Hydroxy Alantolactone from Inula Britannica. Molecules 2017, 22, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, G.-S.; Pae, H.-O.; Jeong, S.-O.; Kim, Y.-C.; Kwon, T.-O.; Lee, H.S.; Kim, N.-S.; Park, S.D.; Chung, H.-T. The Alpha-Methylene-Gamma-Butyrolactone Moiety in Dehydrocostus Lactone Is Responsible for Cytoprotective Heme Oxygenase-1 Expression through Activation of the Nuclear Factor E2-Related Factor 2 in HepG2 Cells. Eur. J. Pharmacol. 2007, 565, 37–44. [Google Scholar] [CrossRef]
- Miyazawa, M.; Shimabayashi, H.; Hayashi, S.; Hashimoto, S.; Nakamura, S.; Kosaka, H.; Kameoka, H. Synthesis and Biological Activity of Alpha-Methylene-Gamma-Lactones as New Aroma Chemicals. J. Agric. Food Chem. 2000, 48, 5406–5410. [Google Scholar] [CrossRef]
- Gach, K.; Janecka, A. α-Methylene-γ-Lactones as a Novel Class of Anti-Leukemic Agents. Anticancer Agents Med. Chem. 2014, 14, 688–694. [Google Scholar] [CrossRef]
- Torres, F.; Quintana, J.; Cabrera, J.; Loro, J.F.; León, F.; Bermejo, J.; Estévez, F. Induction of G2-M Phase Arrest and Apoptosis by Alpha-Methylene-Gamma-Butyrolactones in Human Leukemia Cells. Cancer Lett. 2008, 269, 139–147. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Abraham, A.; Bhat, B.; Jaggi, M.; Singh, A.T.; Sanna, V.K.; Singh, G.; Agarwal, S.K.; Mukherjee, R.; Burman, A.C. Synthesis of 13-Amino Costunolide Derivatives as Anticancer Agents. Bioorg. Med. Chem. Lett. 2006, 16, 4195–4199. [Google Scholar] [CrossRef]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin-From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef]
- FlowJo Software; Version 10.7, Automating HT Flow Cytometry Analysis; BD Biosciences: San Diego, CA, USA, 2014.
- Lim, J.S.; Lee, S.H.; Lee, S.R.; Lim, H.-J.; Roh, Y.-S.; Won, E.J.; Cho, N.; Chun, C.; Cho, Y.-C. Inhibitory Effects of Aucklandia Lappa Decne. Extract on Inflammatory and Oxidative Responses in LPS-Treated Macrophages. Molecules 2020, 25, 1336. [Google Scholar] [CrossRef] [Green Version]
- ImageJ Software; Version 1.52, Image Processing and Analysis in Java; National Institutes of Health: Bethesda, MD, USA, 2018.
- GraphPad Prism Software; Version 5.0, Automating HT Flow Cytometry Analysis; GraphPad, Inc.: San Diego, CA, USA, 2008.






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Cho, Y.-C.; Lim, J.S. Costunolide, a Sesquiterpene Lactone, Suppresses Skin Cancer via Induction of Apoptosis and Blockage of Cell Proliferation. Int. J. Mol. Sci. 2021, 22, 2075. https://doi.org/10.3390/ijms22042075
Lee SH, Cho Y-C, Lim JS. Costunolide, a Sesquiterpene Lactone, Suppresses Skin Cancer via Induction of Apoptosis and Blockage of Cell Proliferation. International Journal of Molecular Sciences. 2021; 22(4):2075. https://doi.org/10.3390/ijms22042075
Chicago/Turabian StyleLee, Sung Ho, Young-Chang Cho, and Jae Sung Lim. 2021. "Costunolide, a Sesquiterpene Lactone, Suppresses Skin Cancer via Induction of Apoptosis and Blockage of Cell Proliferation" International Journal of Molecular Sciences 22, no. 4: 2075. https://doi.org/10.3390/ijms22042075
APA StyleLee, S. H., Cho, Y.-C., & Lim, J. S. (2021). Costunolide, a Sesquiterpene Lactone, Suppresses Skin Cancer via Induction of Apoptosis and Blockage of Cell Proliferation. International Journal of Molecular Sciences, 22(4), 2075. https://doi.org/10.3390/ijms22042075