
Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies

 , ,
, , 
Abstract
:1. Introduction
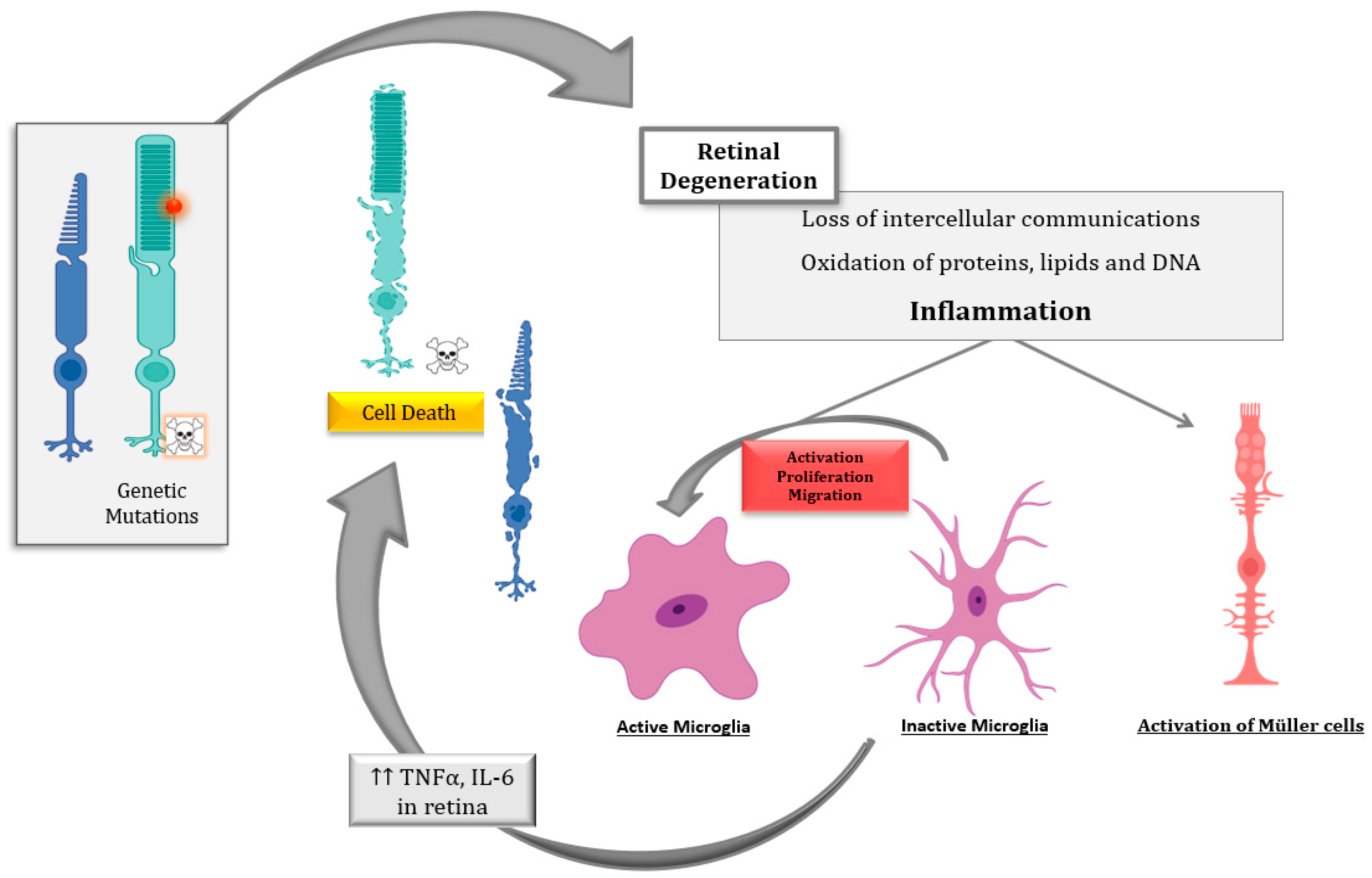
2. Retinal Inflammation in IRDs
2.1. Ocular Inflammation
2.2. Inflammation in RP
2.2.1. Local Inflammation
2.2.2. Peripheral Inflammation
2.3. Inflammation and Cell Death in RP
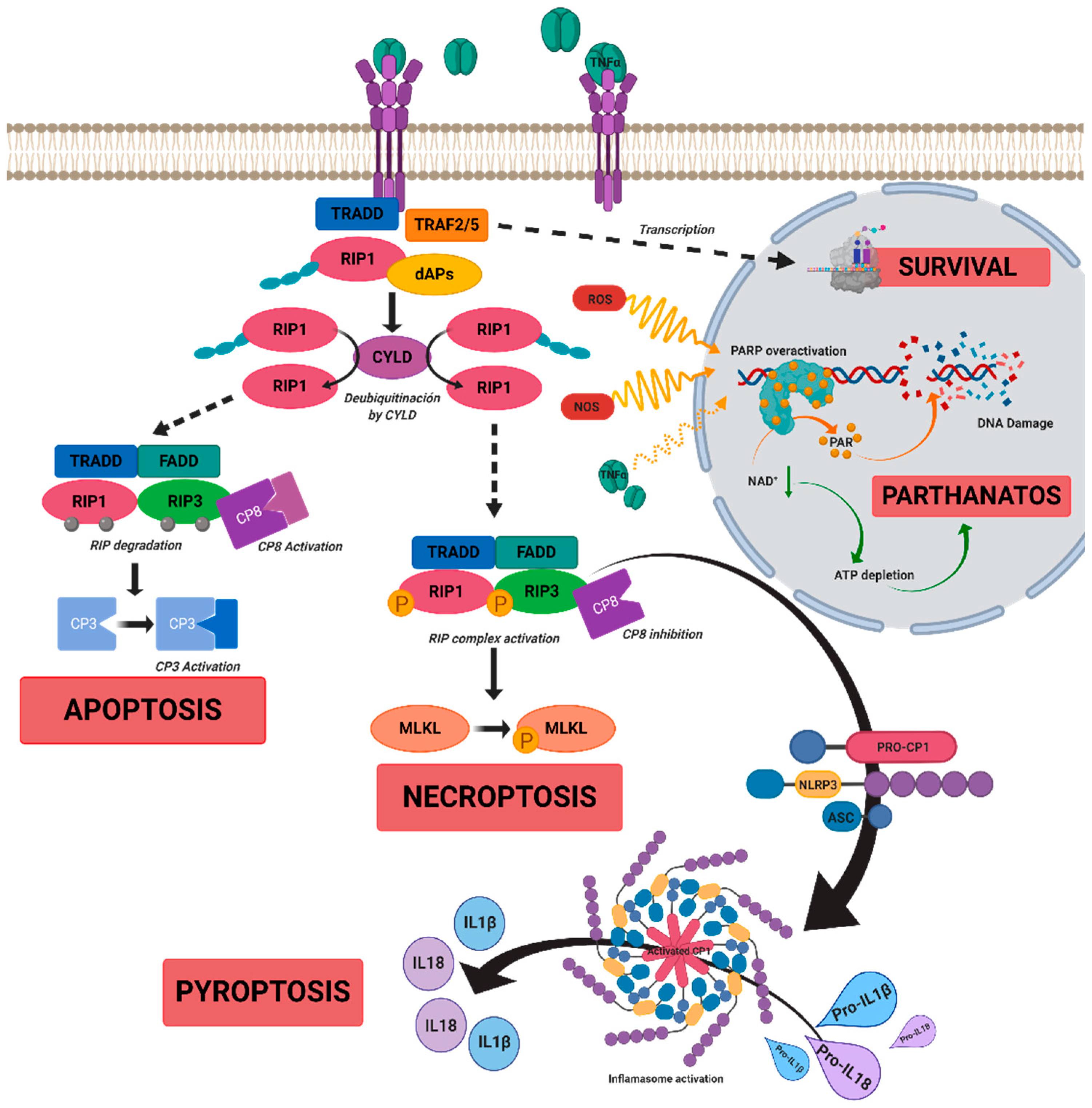
3. Cell Death Mechanisms in IRDs
3.1. Apoptosis
3.2. Necrosis
3.3. Necroptosis
3.4. Pyroptosis
3.5. Parthanatos
4. Therapeutic Approaches to Reduce Inflammation and Cell Death
4.1. Anti-Inflammatory Therapies
4.1.1. Therapies against TNFα
4.1.2. Microglia Inhibition
4.1.3. Other Anti-Inflammatory Strategies
4.2. “Anti-Cell Death” Therapies
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAT | Alpha-1 antitrypsin |
| ADA | Adalimumab |
| ADP-ribose | Adenosine diphosphate ribose |
| AIF | Apoptosis-inducing factor |
| AMD | Age-related macular degeneration |
| cGMP | Cyclic guanosine monophosphate |
| CNS | Central nervous system |
| CPR | C-reactive protein |
| DAMPs | Damage-associated molecular patterns |
| DR | Diabetic retinopathy |
| FADD | Fas-associated protein with death domain |
| GM-CSF | Granulocyte monocyte colony-stimulating factor |
| GSDMD | Gasdermin D |
| HMGB1 | High mobility group box 1 |
| IFN-γ | Gamma interferon |
| IL | Interleukin |
| iNOS | inducible nitric oxide synthase |
| IRDs | Inherited retinal dystrophies |
| JAK-STAT | Janus kinase/signal transducer and activator of transcription |
| M1 | Pro-inflammatory microglia |
| M2 | Anti-inflammatory microglia |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MCP-2 | Monocyte chemoattractant protein 2 |
| MIF | Migration inhibitor factor |
| MAPK | Mitogen-Activated Protein Kinases |
| MLKL | Pseudokinase kinase-like domain of mixed-lineage kinase domain-like |
| NAD+ | Nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor-κappa B |
| ONL | Outer nuclear layer |
| P | Postnatal |
| PAMPs | Pathogen-associated molecular patterns |
| PAR | Poly-ADP ribose |
| PARP | Poly-ADP ribose polymerase |
| PGF | Platelet growth factor |
| RANTES | Regulated upon activation, normal T cell expressed, and secreted |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| RIPK3 | Receptor-interacting serine/threonine-protein kinase 3 |
| ROS | Reactive oxygen species |
| RP | Retinitis pigmentosa |
| TGF-β | Transforming growth factor beta |
| TRAF2 | TNF receptor-associated factor 2 |
| TNFα | Tumor necrosis factor alpha |
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Wooff, Y.; Man, S.M.; Aggio-Bruce, R.; Natoli, R.; Fernando, N. IL-1 Family Members Mediate Cell Death, Inflammation and Angiogenesis in Retinal Degenerative Diseases. Front. Immunol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- RetNet. Retinal Information Network. Available online: https://sph.uth.edu/retnet/home.htm (accessed on 29 December 2020).
- Chang, B. Animal Models of Retinitis Pigmentosa (RP). In Animal Models of Ophthalmic Diseases; Chan, C.-C., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 101–116. [Google Scholar]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Jiao, H.; Natoli, R.; Valter, K.; Provis, J.M.; Rutar, M. Spatiotemporal Cadence of Macrophage Polarisation in a Model of Light-Induced Retinal Degeneration. PLoS ONE 2015, 10, e0143952. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, P.G.; Saban, D.R.; Dando, S.J. Immune cells in the retina and choroid: Two different tissue environments that require different defenses and surveillance. Prog. Retin. Eye Res. 2019, 70, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef]
- Vilhardt, F. Microglia: Phagocyte and glia cell. Int. J. Biochem. Cell Biol. 2005, 37, 17–21. [Google Scholar] [CrossRef]
- Noailles, A.; Fernández-Sánchez, L.; Lax, P.; Cuenca, N. Microglia activation in a model of retinal degeneration and TUDCA neuroprotective effects. J. Neuroinflammation 2014, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Arroba, A.I.; Valverde, Á.M. Modulation of microglia in the retina: New insights into diabetic retinopathy. Acta Diabetol. 2017, 54, 527–533. [Google Scholar] [CrossRef]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.-H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lin, Z.; Liu, C.-H.; Gong, Y.; Liegl, R.; Fredrick, T.W.; Meng, S.S.; Burnim, S.B.; Wang, Z.; Akula, J.D.; et al. Inflammatory signals from photoreceptor modulate pathological retinal angiogenesis via c-Fos. J. Exp. Med. 2017, 214, 1753–1767. [Google Scholar] [CrossRef]
- Massengill, M.T.; Ahmed, C.M.; Lewin, A.S.; Ildefonso, C.J. Neuroinflammation in Retinitis Pigmentosa, Diabetic Retinopathy, and Age-Related Macular Degeneration: A Minireview; Springer International Publishing: Cham, Switzerland, 2018; Volume 1074, pp. 185–191. [Google Scholar]
- Murakami, Y.; Ikeda, Y.; Nakatake, S.; Fujiwara, K.; Tachibana, T.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Yoshida, S.; Ishibashi, T.; et al. C-Reactive protein and progression of vision loss in retinitis pigmentosa. Acta Ophthalmol. 2018, 96, e174–e179. [Google Scholar] [CrossRef] [Green Version]
- Zabel, M.K.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.N.; Wong, W.T. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Newsome, D.A.; Michels, R.G. Detection of Lymphocytes in the Vitreous Gel of Patients with Retinitis Pigmentosa. Am. J. Ophthalmol. 1988, 105, 596–602. [Google Scholar] [CrossRef]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical Evidence of Sustained Chronic Inflammatory Reaction in Retinitis Pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef]
- Appelbaum, T.; Santana, E.; Aguirre, G.D. Strong upregulation of inflammatory genes accompanies photoreceptor demise in canine models of retinal degeneration. PLoS ONE 2017, 12, e0177224. [Google Scholar] [CrossRef] [Green Version]
- Berge, J.C.T.; Fazil, Z.; Born, I.V.D.; Wolfs, R.C.W.; Schreurs, M.W.J.; Dik, W.A.; Rothova, A. Intraocular cytokine profile and autoimmune reactions in retinitis pigmentosa, age-related macular degeneration, glaucoma and cataract. Acta Ophthalmol. 2019, 97, 185–192. [Google Scholar] [CrossRef]
- Okita, A.; Murakami, Y.; Shimokawa, S.; Funatsu, J.; Fujiwara, K.; Nakatake, S.; Koyanagi, Y.; Akiyama, M.; Takeda, A.; Hisatomi, T.; et al. Changes of Serum Inflammatory Molecules and Their Relationships with Visual Function in Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef] [PubMed]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef]
- Mathis, T.; Housset, M.; Eandi, C.; Beguier, F.; Touhami, S.; Reichman, S.; Augustin, S.; Gondouin, P.; Sahel, J.-A.; Kodjikian, L.; et al. Activated monocytes resist elimination by retinal pigment epithelium and downregulate their OTX2 expression via TNF-α. Aging Cell 2017, 16, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Olivares-González, L.; Velasco, S.; Millán, J.M.; Rodrigo, R. Intravitreal administration of adalimumab delays retinal degeneration in rd10 mice. FASEB J. 2020, 34, 13839–13861. [Google Scholar] [CrossRef] [PubMed]
- Cvenkel, B.; Kopitar, A.N.; Ihan, A. Inflammatory Molecules in Aqueous Humour and on Ocular Surface and Glaucoma Surgery Outcome. Mediat. Inflamm. 2010, 2010, 939602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrani, K.; Ahmed, M.; Foster, C.S. Adamantiades-Behcet disease: Diagnosis and current concepts in management of ocular manifestations. Compr. Ophthalmol. Update 2007, 8, 225–233. [Google Scholar]
- Fernández-Vega, B.; Fernández-Vega, Á.; Rangel, C.M.; Nicieza, J.; Villota-Deleu, E.; Vega, J.A.; Sanchez-Avila, R.M. Blockade of Tumor Necrosis Factor-Alpha: A Role for Adalimumab in Neovascular Age-Related Macular Degeneration Refractory to Anti-Angiogenesis Therapy? Case Rep. Ophthalmol. 2016, 7, 154–162. [Google Scholar] [CrossRef]
- Japiassú, R.M.; Brasil, O.F.M.; Cunha, A.L.; De Souza, E.C. Regression of Vasoproliferative Tumor with Systemic Infliximab. Ophthalmic Surg. Lasers Imaging 2008, 39, 348–349. [Google Scholar] [CrossRef]
- Saxena, S.; Khanna, V.K.; Pant, A.B.; Meyer, C.H.; Singh, V.K. Elevated Tumor Necrosis Factor in Serum Is Associated with Increased Retinal Ischemia in Proliferative Eales’ Disease. Pathobiology 2011, 78, 261–265. [Google Scholar] [CrossRef]
- Kocabora, M.S.; Telli, M.E.; Fazil, K.; Erdur, S.K.; Ozsutcu, M.; Çekiç, O.; Ozbilen, K.T. Serum and Aqueous Concentrations of Inflammatory Markers in Diabetic Macular Edema. Ocul. Immunol. Inflamm. 2016, 24, 549–554. [Google Scholar] [CrossRef]
- Sharma, R.K.; Rogojina, A.T.; Chalam, K. Multiplex immunoassay analysis of biomarkers in clinically accessible quantities of human aqueous humor. Mol. Vis. 2009, 15, 60–69. [Google Scholar]
- Takeuchi, M.; Sato, T.; Tanaka, A.; Muraoka, T.; Taguchi, M.; Sakurai, Y.; Karasawa, Y.; Ito, M. Elevated Levels of Cytokines Associated with Th2 and Th17 Cells in Vitreous Fluid of Proliferative Diabetic Retinopathy Patients. PLoS ONE 2015, 10, e0137358. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cámara, C.M.; Hernández-Pinto, A.M.; Olivares-González, L.; Cuevas-Martín, C.; Sánchez-Aragó, M.; Hervás, D.; Salom, D.; Cuezva, J.M.; Enrique, J.; Millán, J.M.; et al. Adalimumab Reduces Photoreceptor Cell Death in A Mouse Model of Retinal Degeneration. Sci. Rep. 2015, 5, 11764. [Google Scholar] [CrossRef] [PubMed]
- de la Cámara, C.M.; Olivares-González, L.; Hervás, D.; Salom, D.; Millán, J.M.; Rodrigo, R. Infliximab reduces Zaprinast-induced retinal degeneration in cultures of porcine retina. J. Neuroinflammation 2014, 11, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares-Gonza´ lez, L.; Martínez-Fernandez de la Ca´ mara, C.; Herva´ s, D.; Mar´ a Milla´ n, J.; Rodrigo, R. HIF-1α stabilization reduces retinal degeneration in a mouse model of retinitis pigmentosa. FASEB J. 2018, 32, 2438–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, T.; Kotla, P.; Fullard, R.; Gorbatyuk, M. TNFa knockdown in the retina promotes cone survival in a mouse model of autosomal dominant retinitis pigmentosa. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 92–102. [Google Scholar] [CrossRef]
- Niederkorn, J.Y. Immune escape mechanisms of intraocular tumors. Prog. Retin. Eye Res. 2009, 28, 329–347. [Google Scholar] [CrossRef] [Green Version]
- Noailles, A.; Maneu, V.; Campello, L.; Lax, P.; Cuenca, N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis. 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Xiao, J.; Wang, K.; So, K.-F.; Tipoe, G.L.; Lin, B. Suppression of Microglial Activation Is Neuroprotective in a Mouse Model of Human Retinitis Pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef] [Green Version]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.L. Mechanisms of Photoreceptor Death During Retinal Degeneration. Optom. Vis. Sci. 2010, 87, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Silva-Vilches, C.; Ring, S.; Mahnke, K. ATP and Its Metabolite Adenosine as Regulators of Dendritic Cell Activity. Front. Immunol. 2018, 9, 2581. [Google Scholar] [CrossRef]
- Wooff, Y.; Fernando, N.; Wong, J.H.C.; Dietrich, C.; Aggio-Bruce, R.; Chu-Tan, J.A.; Robertson, A.A.B.; Doyle, S.L.; Man, S.M.; Natoli, R. Caspase-1-dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration. Sci. Rep. 2020, 10, 2263. [Google Scholar] [CrossRef] [Green Version]
- Viringipurampeer, I.A.; Metcalfe, A.L.; Bashar, A.E.; Sivak, O.; Yanai, A.; Mohammadi, Z.; Moritz, O.L.; Gregory-Evans, C.Y.; Gregory-Evans, K. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum. Mol. Genet. 2016, 25, 1501–1516. [Google Scholar] [CrossRef] [Green Version]
- Sudharsan, R.; Beiting, D.P.; Aguirre, G.D.; Beltran, W.A. Involvement of Innate Immune System in Late Stages of Inherited Photoreceptor Degeneration. Sci. Rep. 2017, 7, 17897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.-Q.; Hao, Y.; Wong, F. Apoptosis: Final common pathway of photoreceptor death in rd, rds, and mutant mice. Neuron 1993, 11, 595–605. [Google Scholar] [CrossRef]
- Viringipurampeer, I.A.; Gregory-Evans, C.Y.; Metcalfe, A.L.; Bashar, E.; Moritz, O.L.; Gregory-Evans, K. Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Mol. Neurobiol. 2019, 56, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zhang, H.; He, D.; Wang, Y.; Cai, B.; Chen, J.; Ma, J.; Liu, Z.; Wu, Y. Retinal Pigment Epithelium Cell Death Is Associated with NLRP3 Inflammasome Activation by All-trans Retinal. Investig. Opthalmology Vis. Sci. 2019, 60, 3034–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Cui, J.Z.; To, E.; Cao, S.; Matsubara, J.A. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J. Neuroinflammation 2018, 15, 15. [Google Scholar] [CrossRef]
- Power, M.; Das, S.; Schütze, K.; Marigo, V.; Ekström, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin. Eye Res. 2020, 74, 100772. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Trachsel-Moncho, L.; Benlloch-Navarro, S.; Fernández-Carbonell, Á.; Ramírez-Lamelas, D.T.; Olivar, T.; Silvestre, D.; Poch, E.; Miranda, M. Oxidative stress and autophagy-related changes during retinal degeneration and development. Cell Death Dis. 2018, 9, 812. [Google Scholar] [CrossRef] [Green Version]
- Nirmala, J.G.; Lopus, M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2020, 36, 145–164. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Bhootada, Y.; Gorbatyuk, M.S. Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis. 2013, 4, e528. [Google Scholar] [CrossRef] [Green Version]
- Comitato, A.; Sanges, D.; Rossi, A.; Humphries, M.M.; Marigo, V. Activation of Bax in Three Models of Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3555–3561. [Google Scholar] [CrossRef] [Green Version]
- Kunte, M.M.; Choudhury, S.; Manheim, J.F.; Shinde, V.M.; Miura, M.; Chiodo, V.A.; Hauswirth, W.W.; Gorbatyuk, O.S.; Gorbatyuk, M.S. ER Stress Is Involved in T17M Rhodopsin-Induced Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3792–3800. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Wang, Z.-J. (Z)-7,4’-Dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside mitigates retinal degeneration in Rd10 mouse model through inhibiting oxidative stress and inflammatory responses. Cutan. Ocul. Toxicol. 2020, 39, 36–42. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Peng, M.; Laties, A.M.; Wen, R. Activation of Caspase-3 in the Retina of Transgenic Rats with the Rhodopsin Mutation S334ter during Photoreceptor Degeneration. J. Neurosci. 1999, 19, 4778–4785. [Google Scholar] [CrossRef] [Green Version]
- Zeiss, C.J.; Neal, J.; Johnson, E.A. Caspase-3 in postnatal retinal development and degeneration. Investig. Ophthalmol. Vis. Sci. 2004, 45, 964–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.J.; Song, W.T.; Yao, F.; Zhang, X.; Peng, J.; Luo, X.J.; Xia, X.B. Involvement of regulated necrosis in blinding diseases: Focus on necroptosis and ferroptosis. Exp. Eye Res. 2020, 191, 107922. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; Eldeiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Berghe, T.V. The Role of the Kinases RIP1 and RIP3 in TNF-Induced Necrosis. Sci. Signal. 2010, 3, re4. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Spall, S.K.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.-L.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Suzuki, J.; Hisatomi, T.; Ikeda, Y.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 14598–14603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hachim, M.Y.; Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Pyroptosis: The missing puzzle among innate and adaptive immunity crosstalk. J. Leukoc. Biol. 2020, 108, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Genet. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef]
- Kaur, J.; Mencl, S.; Sahaboglu, A.; Farinelli, P.; Van Veen, T.; Zrenner, E.; Ekström, P.; Paquet-Durand, F.; Arango-Gonzalez, B. Calpain and PARP Activation during Photoreceptor Cell Death in P23H and S334ter Rhodopsin Mutant Rats. PLoS ONE 2011, 6, e22181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahaboglu, A.; Miranda, M.; Canjuga, D.; Avci-Adali, M.; Savytska, N.; Secer, E.; Feria-Pliego, J.A.; Kayık, G.; Durdagi, S. Drug repurposing studies of PARP inhibitors as a new therapy for inherited retinal degeneration. Cell. Mol. Life Sci. 2020, 77, 2199–2216. [Google Scholar] [CrossRef]
- Jayakody, S.A.; Gonzalez-Cordero, A.; Ali, R.R.; Pearson, R.A. Cellular strategies for retinal repair by photoreceptor replacement. Prog. Retin. Eye Res. 2015, 46, 31–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamam, R.N.; Barikian, A.W.; Antonios, R.S.; Abdulaal, M.R.; Alameddine, R.M.; El Mollayess, G.; Mansour, A.M. Intravitreal Adalimumab in Active Noninfectious Uveitis: A Pilot Study. Ocul. Immunol. Inflamm. 2016, 24, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Mirshahi, A.; Hoehn, R.; Lorenz, K.; Kramann, C.; Baatz, H. Anti-Tumor Necrosis Factor Alpha for Retinal Diseases: Current Knowledge and Future Concepts. J. Ophthalmic Vis. Res. 2012, 7, 39–44. [Google Scholar]
- Wu, L.; Arevalo, J.F.; Hernandez-Bogantes, E.; Regatieri, C.V.; Roca, J.A.; Farah, M.E. Intravitreal Tumor Necrosis Factor-Alpha Inhibitors for Neovascular Age-Related Macular Degeneration Suboptimally Responsive to Antivascular Endothelial Growth Factor Agents: A Pilot Study from the Pan American Collaborative Retina Study Group. J. Ocul. Pharmacol. Ther. 2013, 29, 366–371. [Google Scholar] [CrossRef]
- Wu, L.; Hernandez-Bogantes, E.; Roca, J.A.; Arevalo, J.F.; Barraza, K.; Lasave, A.F. Intravitreal tumor necrosis factor inhibitors in the treatment of refractory diabetic macular edema: A pilot study from the Pan-American Collaborative Retina Study Group. Retina 2011, 31, 298–303. [Google Scholar] [CrossRef]
- Llorenç, V.; Mesquida, M.; De La Maza, M.S.; Blanco, R.; Calvo, V.; Maíz, O.; Blanco, A.; Aberásturi, J.R.D.D.-J.D.; Adán, A. Certolizumab Pegol, a New Anti-TNF-α in the Armamentarium against Ocular Inflammation. Ocul. Immunol. Inflamm. 2016, 24, 167–172. [Google Scholar] [CrossRef]
- Hasegawa, E.; Takeda, A.; Yawata, N.; Sonoda, K.-H. The effectiveness of adalimumab treatment for non-infectious uveitis. Immunol. Med. 2019, 42, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Lichtlen, P.; Lam, T.T.; Nork, T.M.; Streit, T.; Urech, D.M. Relative Contribution of VEGF and TNF-α in the Cynomolgus Laser-Induced CNV Model: Comparing the Efficacy of Bevacizumab, Adalimumab, and ESBA105. Investig. Opthalmology Vis. Sci. 2010, 51, 4738–4745. [Google Scholar] [CrossRef]
- Joussen, A.M.; Doehmen, S.; Le, M.L.; Koizumi, K.; Radetzky, S.; Krohne, T.U.; Poulaki, V.; Semkova, I.; Kociok, N. TNF-α mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol. Vis. 2009, 15, 1418–1428. [Google Scholar] [PubMed]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a Widely Used Inhibitor of Tumor Necrosis Factor-α (TNF- α), Prevents Retinal Ganglion Cell Loss in a Rat Model of Glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schick, T.; Steinhauer, M.; Aslanidis, A.; Altay, L.; Karlstetter, M.; Langmann, T.; Kirschfink, M.; Fauser, S. Local complement activation in aqueous humor in patients with age-related macular degeneration. Eye 2017, 31, 810–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, M.H.; Rashid, K.; Ambrósio, A.F.; Santiago, A.R.; Langmann, T. Blockade of microglial adenosine A2A receptor impacts inflammatory mechanisms, reduces ARPE-19 cell dysfunction and prevents photoreceptor loss in vitro. Sci. Rep. 2018, 8, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.C.; Allen, D.M.; Cardona, A.E. The Role of Microglia in Diabetic Retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.-Y.; Green, W.R.; Tso, M.O.M. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.-L.; Shi, J.-M. The role of microglia in the progression of glaucomatous neurodegeneration—A review. Int. J. Ophthalmol. 2018, 11, 143–149. [Google Scholar] [CrossRef]
- Arroba, A.I.; Campos-Caro, A.; Aguilar-Diosdado, M.; Valverde, Á.M. IGF-1, Inflammation and Retinal Degeneration: A Close Network. Front. Aging Neurosci. 2018, 10, 203. [Google Scholar] [CrossRef]
- Arroba, A.I.; Alcalde-Estevez, E.; García-Ramírez, M.; Cazzoni, D.; De La Villa, P.; Sánchez-Fernández, E.M.; Mellet, C.O.; Fernández, J.M.G.; Hernández, C.; Simó, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1663–1674. [Google Scholar] [CrossRef]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline Reduces Proinflammatory Cytokine Expression, Microglial Activation, and Caspase-3 Activation in a Rodent Model of Diabetic Retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, Y.; Wu, Q.; Jia, L.; Du, X. Minocycline inhibits PARP-1 expression and decreases apoptosis in diabetic retinopathy. Mol. Med. Rep. 2015, 12, 4887–4894. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced Retina Microglial Activation and Improved Optic Nerve Integrity with Minocycline Treatment in the DBA/2J Mouse Model of Glaucoma. Investig. Opthalmology Vis. Sci. 2008, 49, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Benlloch-Navarro, S.; Trachsel-Moncho, L.; Fernández-Carbonell, Á.; Olivar, T.; Soria, J.M.; Almansa, I.; Miranda, M. Progesterone anti-inflammatory properties in hereditary retinal degeneration. J. Steroid Biochem. Mol. Biol. 2019, 189, 291–301. [Google Scholar] [CrossRef]
- Roche, S.L.; Ruiz-Lopez, A.M.; Moloney, J.N.; Byrne, A.M.; Cotter, T.G. Microglial-induced Müller cell gliosis is attenuated by progesterone in a mouse model of retinitis pigmentosa. Glia 2018, 66, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.L.; Kutsyr, O.; Cuenca, N.; Cotter, T.G. Norgestrel, a Progesterone Analogue, Promotes Significant Long-Term Neuroprotection of Cone Photoreceptors in a Mouse Model of Retinal Disease. Investig. Opthalmology Vis. Sci. 2019, 60, 3221–3235. [Google Scholar] [CrossRef]
- Guadagni, V.; Biagioni, M.; Novelli, E.; Aretini, P.; Mazzanti, C.M.; Strettoi, E. Rescuing cones and daylight vision in retinitis pigmentosa mice. FASEB J. 2019, 33, 10177–10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Dong, X.; Lu, X.; Qu, Y.; Wang, C.; Peng, G.; Zhang, J. Subcutaneous delivery of tauroursodeoxycholic acid rescues the cone photoreceptors in degenerative retina: A promising therapeutic molecule for retinopathy. Biomed. Pharmacother. 2019, 117, 109021. [Google Scholar] [CrossRef]
- Ramírez-Lamelas, D.T.; Benlloch-Navarro, S.; Lopez-Pedrajas, R.; Gimeno-Hernández, R.; Olivar, T.; Silvestre, D.; Miranda, M. Lipoic Acid and Progesterone Alone or in Combination Ameliorate Retinal Degeneration in an Experimental Model of Hereditary Retinal Degeneration. Front. Pharmacol. 2018, 9, 469. [Google Scholar] [CrossRef] [Green Version]
- Terluk, M.R.; Ebeling, M.C.; Fisher, C.R.; Kapphahn, R.J.; Yuan, C.; Kartha, R.V.; Montezuma, S.R.; Ferrington, D.A. N-Acetyl-L-cysteine Protects Human Retinal Pigment Epithelial Cells from Oxidative Damage: Implications for Age-Related Macular Degeneration. Oxidative Med. Cell. Longev. 2019, 2019, 5174957-14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yin, Z.; Gao, L.; Sun, D.; Hu, X.; Xue, L.; Dai, J.; Zeng, Y.; Chen, S.; Pan, B.; et al. Curcumin Delays Retinal Degeneration by Regulating Microglia Activation in the Retina of rd1 Mice. Cell. Physiol. Biochem. 2017, 44, 479–493. [Google Scholar] [CrossRef]
- Chan, H.H.-L.; Lam, H.-I.; Choi, K.-Y.; Li, S.Z.-C.; Lakshmanan, Y.; Yu, W.-Y.; Chang, R.C.-C.; Lai, J.S.-M.; So, K.-F. Delay of cone degeneration in retinitis pigmentosa using a 12-month treatment with Lycium barbarum supplement. J. Ethnopharmacol. 2019, 236, 336–344. [Google Scholar] [CrossRef]
- Tang, L.; Bao, S.; Du, Y.; Jiang, Z.; Wuliji, A.; Ren, X.; Zhang, C.; Chu, H.; Kong, L.; Ma, H. Antioxidant effects of Lycium barbarum polysaccharides on photoreceptor degeneration in the light-exposed mouse retina. Biomed. Pharmacother. 2018, 103, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, J.; Rashid, K.; Langmann, T. Resveratrol induces dynamic changes to the microglia transcriptome, inhibiting inflammatory pathways and protecting against microglia-mediated photoreceptor apoptosis. Biochem. Biophys. Res. Commun. 2018, 501, 239–245. [Google Scholar] [CrossRef]
- Lew, D.S.; Mazzoni, F.; Finnemann, S.C. Microglia Inhibition Delays Retinal Degeneration Due to MerTK Phagocytosis Receptor Deficiency. Front. Immunol. 2020, 11, 1463. [Google Scholar] [CrossRef]
- Chumsakul, O.; Wakayama, K.; Tsuhako, A.; Baba, Y.; Takai, Y.; Kurose, T.; Honma, Y.; Watanabe, S. Apigenin Regulates Activation of Microglia and Counteracts Retinal Degeneration. J. Ocul. Pharmacol. Ther. 2020, 36, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, Z.; Zhu, X.; Sun, X.; Liu, Y.; Cheng, B.; Li, M.; Liu, Y.; He, C.; Liu, X. Alpha-1 Antitrypsin Attenuates M1 Microglia-Mediated Neuroinflammation in Retinal Degeneration. Front. Immunol. 2018, 9, 1202. [Google Scholar] [CrossRef]
- Ahn, S.J.; Kim, K.E.; Woo, S.J.; Park, K.H.; Joon, A.S.; Eun, K.K.; Hyung, P.K. The Effect of an Intravitreal Dexamethasone Implant for Cystoid Macular Edema in Retinitis Pigmentosa: A Case Report and Literature Review. Ophthalmic Surg. Lasers Imaging Retin. 2014, 45, 160–164. [Google Scholar] [CrossRef]
- Zhang, X.; Shahani, U.; Reilly, J.; Shu, X. Disease mechanisms and neuroprotection by tauroursodeoxycholic acid in Rpgr knockout mice. J. Cell. Physiol. 2019, 234, 18801–18812. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rabaza, V.; López-Pedrajas, R.; Almansa, I. Progesterone, Lipoic Acid, and Sulforaphane as Promising Antioxidants for Retinal Diseases: A Review. Antioxidants 2019, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Usui, S.; Zafar, A.-B.; Oveson, B.C.; Jo, Y.-J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell. Physiol. 2011, 226, 1843–1849. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, J.; Xiang, Z.; Xu, D.; So, K.-F.; Vardi, N.; Xu, Y. Lycium Barbarum Polysaccharides Protect Retina in rd1 Mice During Photoreceptor Degeneration. Investig. Opthalmology Vis. Sci. 2018, 59, 597–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campochiaro, P.A.; Iftikhar, M.; Hafiz, G.; Akhlaq, A.; Tsai, G.; Wehling, D.; Lu, L.; Wall, G.M.; Singh, M.S.; Kong, X. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Investig. 2020, 130, 1527–1541. [Google Scholar] [CrossRef] [Green Version]
- Vasireddy, V.; Chavali, V.R.M.; Joseph, V.T.; Kadam, R.; Lin, J.H.; Jamison, J.A.; Kompella, U.B.; Reddy, G.B.; Ayyagari, R. Rescue of Photoreceptor Degeneration by Curcumin in Transgenic Rats with P23H Rhodopsin Mutation. PLoS ONE 2011, 6, e21193. [Google Scholar] [CrossRef] [Green Version]
- Scott, P.A.; Kaplan, H.J.; McCall, M.A. Prenatal Exposure to Curcumin Protects Rod Photoreceptors in a Transgenic Pro23His Swine Model of Retinitis Pigmentosa. Transl. Vis. Sci. Technol. 2015, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Leonard, K.C.; Petrin, D.; Coupland, S.G.; Baker, A.N.; Leonard, B.C.; Lacasse, E.C.; Hauswirth, W.W.; Korneluk, R.G.; Tsilfidis, C. XIAP Protection of Photoreceptors in Animal Models of Retinitis Pigmentosa. PLoS ONE 2007, 2, e314. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Sanges, D.; McCall, J.; Silva, J.; Van Veen, T.; Marigo, V.; Ekström, P. Photoreceptor rescue and toxicity induced by different calpain inhibitors. J. Neurochem. 2010, 115, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.R.; Kim, J.H.; Park, H.Y.; Park, C.K. Ischemia Reperfusion Injury Triggers TNFalpha Induced-Necroptosis in Rat Retina. Curr. Eye Res. 2017, 42, 771–779. [Google Scholar] [CrossRef]
- Sato, K.; Li, S.; Gordon, W.C.; He, J.; Liou, G.I.; Hill, J.M.; Travis, G.H.; Bazan, N.G.; Jin, M. Receptor Interacting Protein Kinase-Mediated Necrosis Contributes to Cone and Rod Photoreceptor Degeneration in the Retina Lacking Interphotoreceptor Retinoid-Binding Protein. J. Neurosci. 2013, 33, 17458–17468. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Sahaboglu, A.; Zrenner, E.; Ueffing, M.; Ekström, P.A.R.; Paquet-Durand, F. Efficacy of PARP inhibition in Pde6a mutant mouse models for retinitis pigmentosa depends on the quality and composition of individual human mutations. Cell Death Discov. 2016, 2, 16040. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Barth, M.; Secer, E.; Del Amo, E.M.; Urtti, A.; Arsenijevic, Y.; Zrenner, E.; Paquet-Durand, F. Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep. 2016, 6, 39537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifunović, D.; Petridou, E.; Comitato, A.; Marigo, V.; Ueffing, M.; Paquet-Durand, F. Primary Rod and Cone Degeneration Is Prevented by HDAC Inhibition. Adv. Exp. Med. Biol. 2018, 1074, 367–373. [Google Scholar] [CrossRef]
- Yao, J.; Qiu, Y.; Frontera, E.; Jia, L.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Thompson, D.A.; Zacks, D.N. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 2018, 14, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, B.; Sun, Y.-J.; Liu, S.-Y.; Che, L.; Li, G.-Y. Neuroprotective Strategy in Retinal Degeneration: Suppressing ER Stress-Induced Cell Death via Inhibition of the mTOR Signal. Int. J. Mol. Sci. 2017, 18, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Compound | Effect | Model of Degeneration or Patients | Disease | References |
|---|---|---|---|---|
| Adalimumab | Reduction of photoreceptor cell death, NLRP3 inflammasome components, PARP activation and microglial activation. | rd10 mice | RP | [28] |
| Steroid hormones Progesterone or norgestrel | Decrease of rod and cone cell death, reactive gliosis, Activation of microglia, and release of inflammatory factors. | rd10 mice | RP | [105,106,107] |
| Dexamethasone | Decrease of inflammation, increase of cone survival, and improvement of visual acuity. | rd10 mice | RP | [108] |
| TUDCA | Reduction of photoreceptor degeneration, inflammation, and microglia activation, and improvement of visual function. | Rpgr knockout mice; mice with N-methyl-N-nitrosourea | RP, retinal degeneration | [109] |
| Lipoic acid | Decrease of photoreceptor cell death and microglial activation. | rd1 mice | RP | [110] |
| N-acetylcysteine | Reduction in cone cell death and improvement in cone function Suppression of inflammatory factors and microglial activation. | rd1 and rd10 mice RP patients | RP | [111] |
| Turmeric (curcumin) | Decreased number of apoptotic cells, improvement of visual function, inhibition of microglial activation, and secretion of chemokines. | rd1 mice, 661W and BV2 cells | RP | [112] |
| Lycium barbarum (Goji berries) | Promotion of photoreceptor survival, improvement of retinal morphology, improvement in visual function; reduction of oxidative stress through the regulation of antioxidant genes. | rd1 mice RP patients | RP | [113,114] |
| Resveratrol | Blockade of expression of pro-inflammatory molecules (IL-1β and IL-6) and protection against photoreceptor apoptosis | 661W and BV2 cells | Retinal degeneration | [115] |
| Tamoxifen and liposomal clodronate | Reduction of photoreceptor death by suppressing microglia activation. | RCS mutMerTK-RP rat | RP | [116] |
| Minocycline | Reduction of photoreceptor cell death and improvement of retinal structure and function. | rd10 mice | RP | [45] |
| Apigenin | Reduction of expression of inflammatory chemokines and oxidative stress; suppression of microglia and Müller glia activation; increase in the thickness of the photoreceptor layer. | rd1 mice | RP | [117] |
| Alpha-1 antitrypsin (AAT) | Attenuation of neuroinflammation by improving visual function and alleviating photoreceptor degeneration. | rd1 mice | RP | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. https://doi.org/10.3390/ijms22042096
Olivares-González L, Velasco S, Campillo I, Rodrigo R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. International Journal of Molecular Sciences. 2021; 22(4):2096. https://doi.org/10.3390/ijms22042096
Chicago/Turabian StyleOlivares-González, Lorena, Sheyla Velasco, Isabel Campillo, and Regina Rodrigo. 2021. "Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies" International Journal of Molecular Sciences 22, no. 4: 2096. https://doi.org/10.3390/ijms22042096