
Structural Insights into Carboxylic Polyester-Degrading Enzymes and Their Functional Depolymerizing Neighbors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Structure Comparison
2.2. Residue Co-Evolution
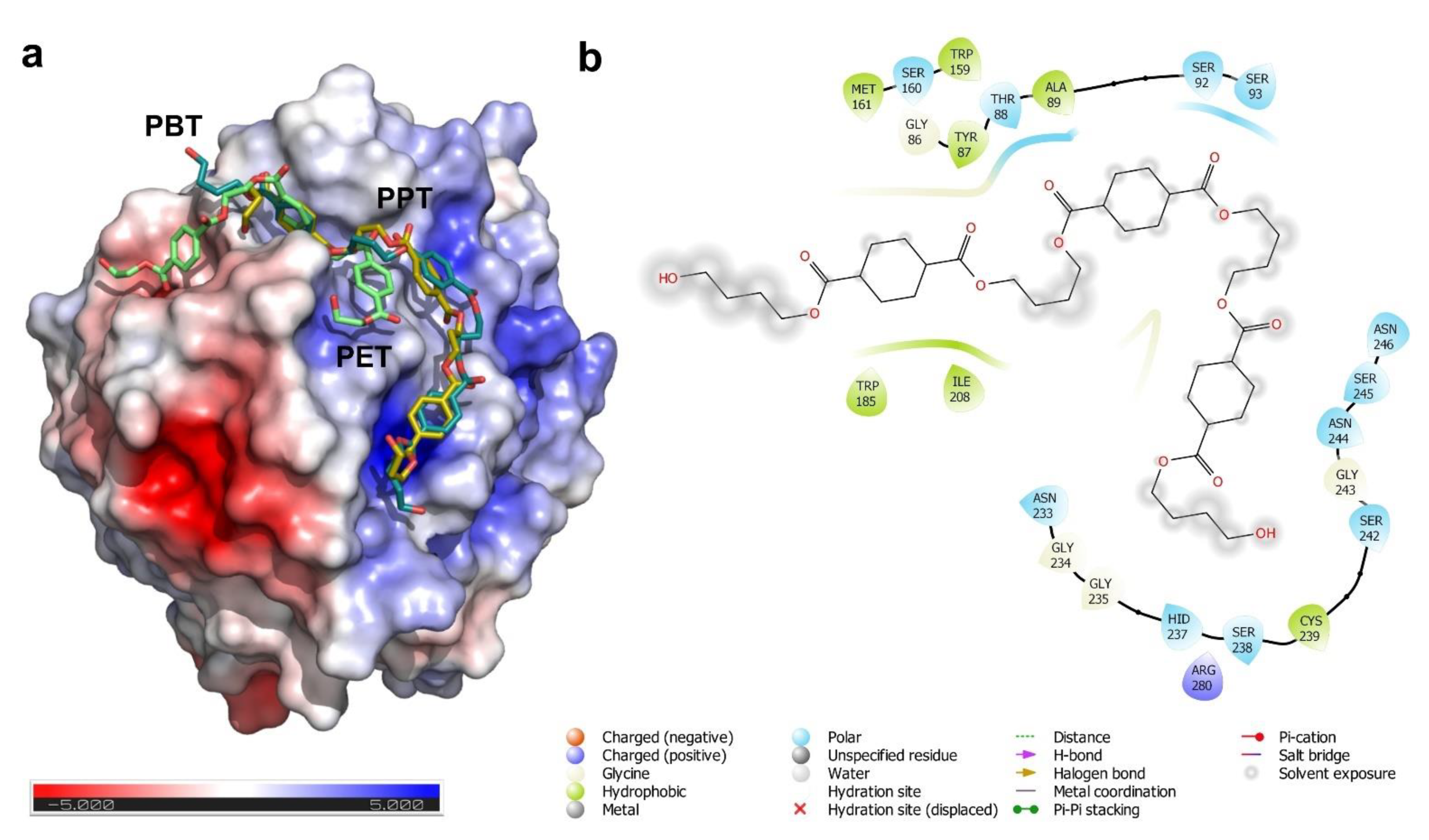
2.3. Ligand Docking
2.4. Data Representation and Analysis
3. Results
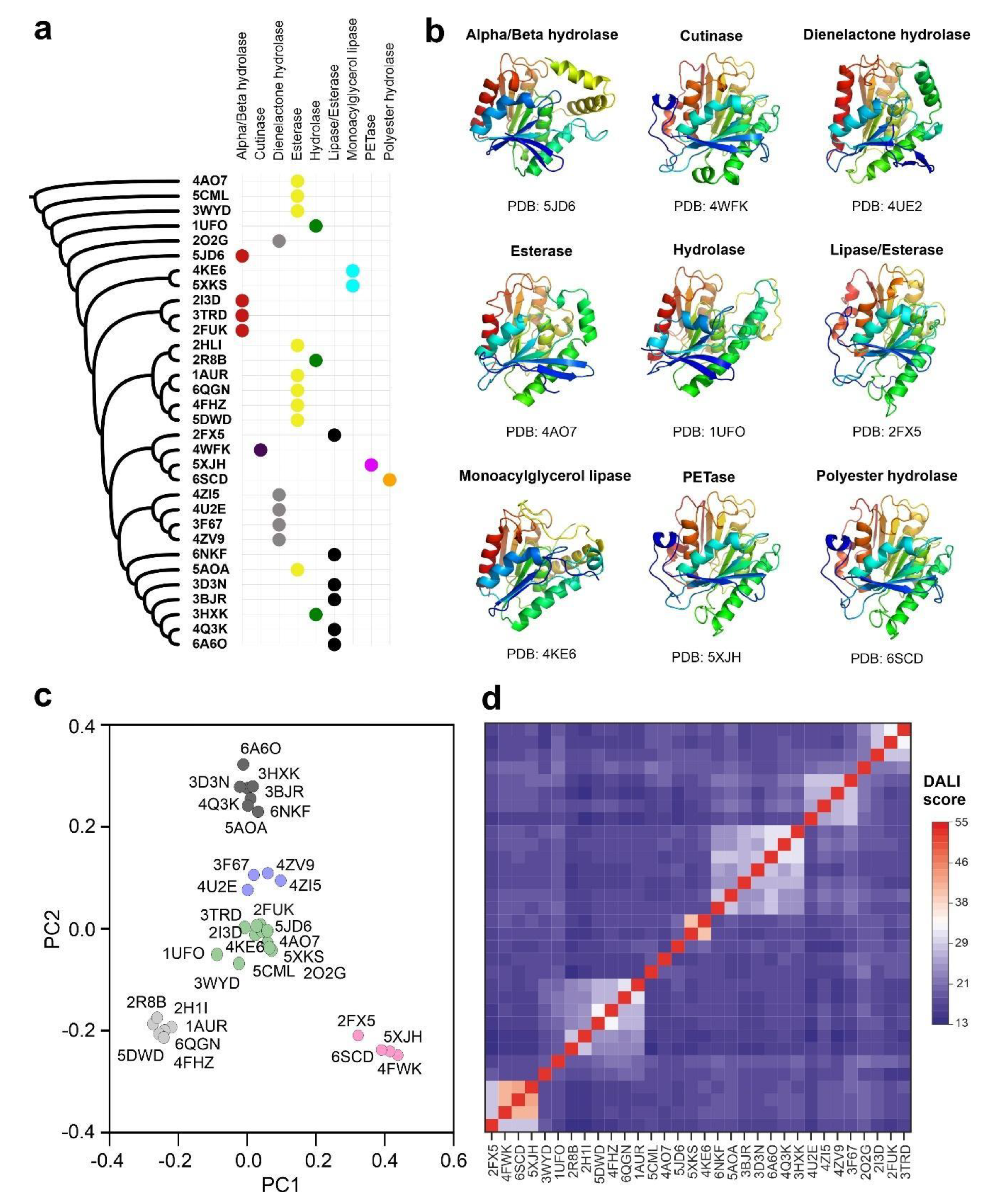
3.1. Carboxyl-Esterase Fold Is Widespread in Prokaryotic and Eukaryotic Proteomes
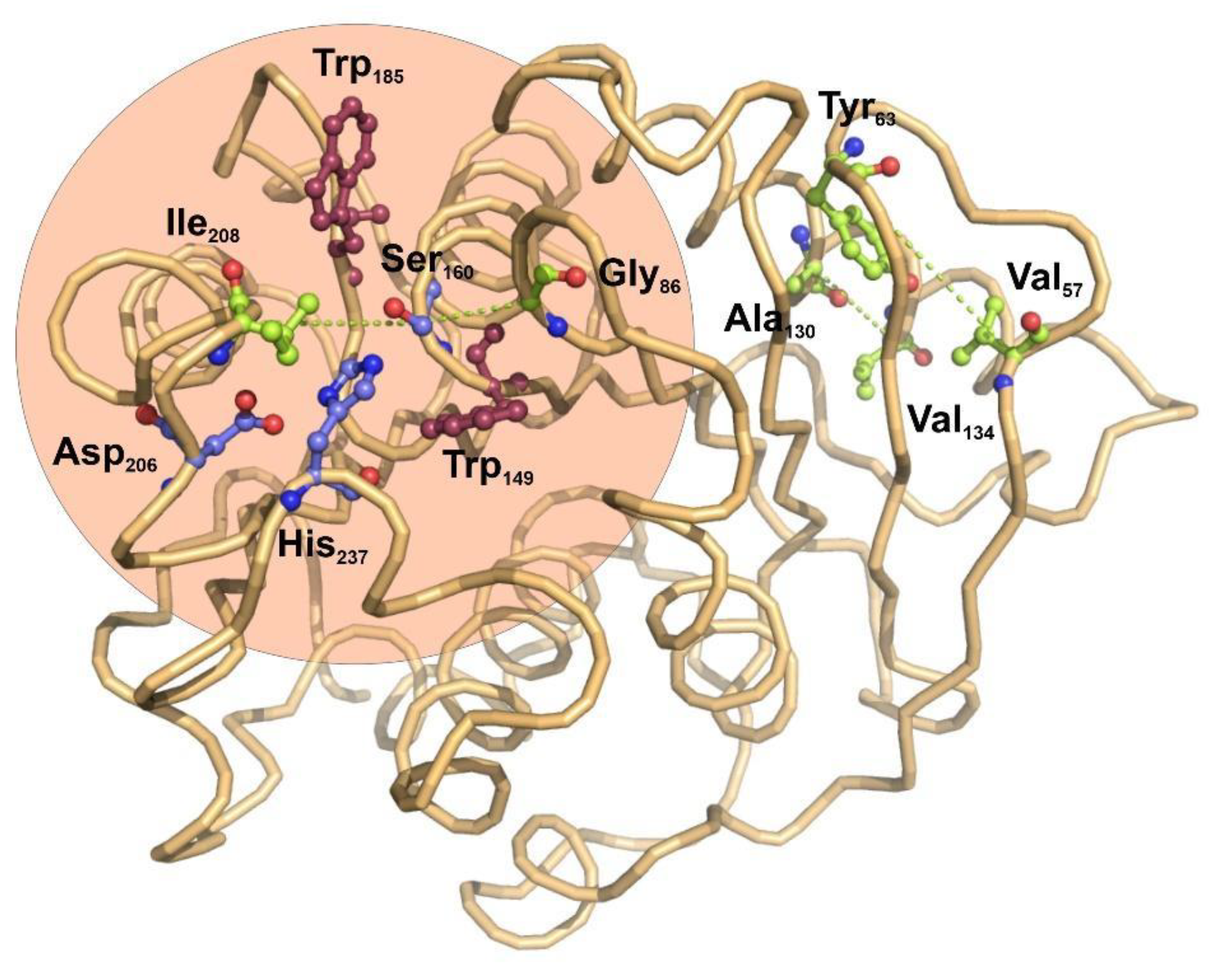
3.2. Coevolution of Catalytic and Structural Residues in Polyester-Hydrolases
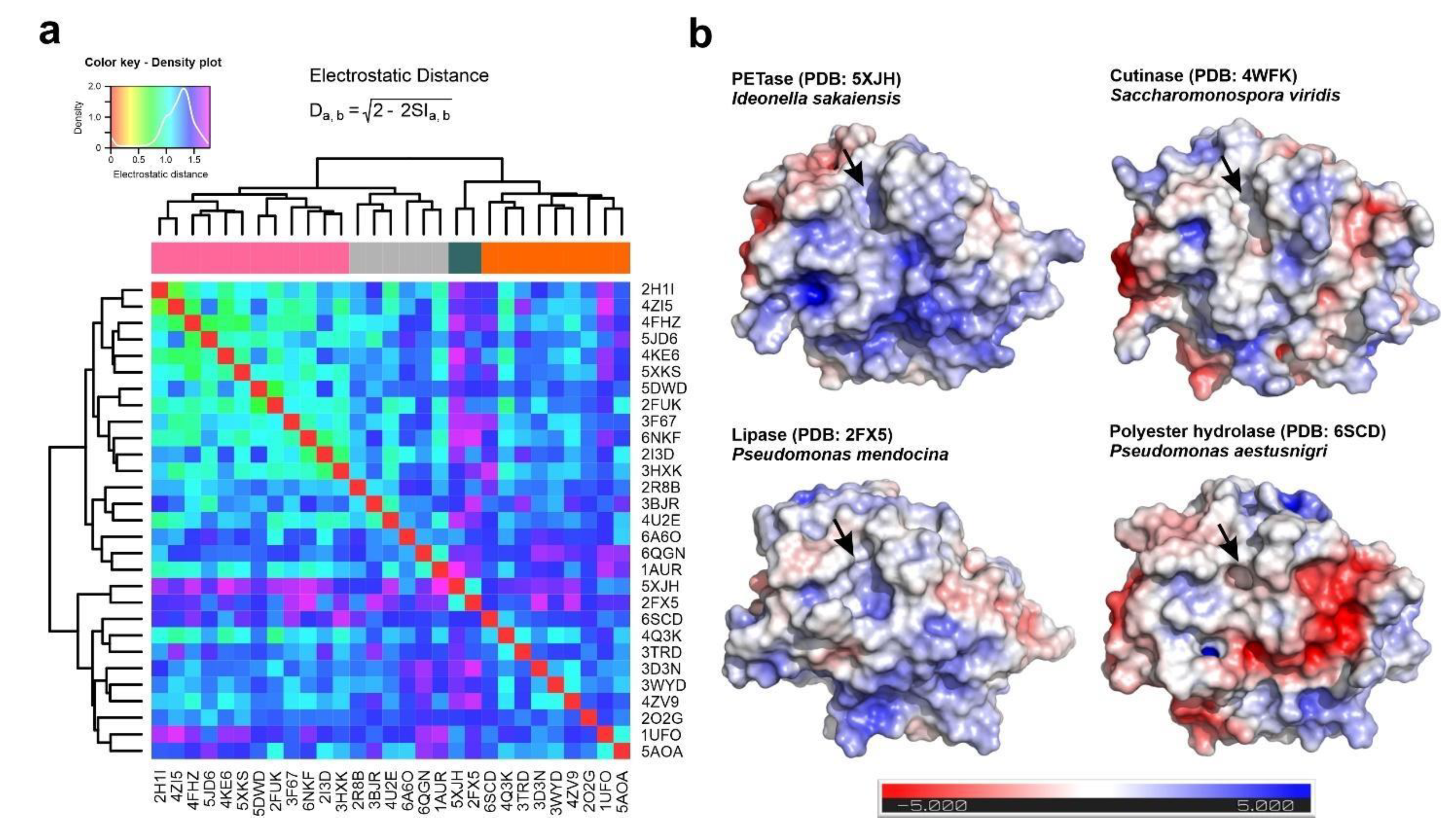
3.3. Substrate Binding and Surface Electrostatics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simons, K.; Vaz, W.L. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef]
- Heredia, A. Biophysical and biochemical characteristics of cutin, a plant barrier biopolymer. Biochim. Biophys. Acta 2003, 1620, 1–7. [Google Scholar] [CrossRef]
- Li, Z.; Hong, H.; Wang, S.-h. Fuzzy-integrative judgment on the end-use performance of knitted fabrics made with polytrimethylene terephthalate blended yarns. Text. Res. J. 2011, 81, 1739–1747. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Fangteng, J.; Wang, S.; Fang, L.; Shen, H.; Xiang, S.; Sun, H.; Yang, B. Bioinspired polyethylene terephthalate nanocone arrays with underwater superoleophobicity and anti-bioadhesion properties. Nanoscale 2014, 6, 13845–13853. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.S.; Islam, M.N.; Rahman, M.M.; Hannan, M.O.; Dungani, R.; Khalil, H.A. Flat-pressed wood plastic composites from sawdust and recycled polyethylene terephthalate (PET): Physical and mechanical properties. Springerplus 2013, 2, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerhoff, P.; Prapaipong, P.; Shock, E.; Hillaireau, A. Antimony leaching from polyethylene terephthalate (PET) plastic used for bottled drinking water. Water Res. 2008, 42, 551–556. [Google Scholar] [CrossRef]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambarini, V.; Pantos, O.; Kingsbury, J.M.; Weaver, L.; Handley, K.M.; Lear, G. Phylogenetic Distribution of Plastic-Degrading Microorganisms. mSystems 2021, 6. [Google Scholar] [CrossRef]
- Tanasupawat, S.; Takehana, T.; Yoshida, S.; Hiraga, K.; Oda, K. Ideonella sakaiensis sp. nov., isolated from a microbial consortium that degrades poly(ethylene terephthalate). Int. J. Syst. Evol. Microbiol. 2016, 66, 2813–2818. [Google Scholar] [CrossRef] [PubMed]
- Tacin, M.V.; Costa-Silva, T.A.; de Paula, A.V.; Palomo, J.M.; Santos-Ebinuma, V.C. Microbial lipase: A new approach for a heterogeneous biocatalyst. Prep. Biochem. Biotechnol. 2020, 1–12. [Google Scholar] [CrossRef]
- Sood, S.; Sharma, A.; Sharma, N.; Kanwar, S.S. Carboxylesterases: Sources, Characterization and Broader Applications. Insights Enzym. Res. 2018, 1. [Google Scholar] [CrossRef]
- Rauwerdink, A.; Lunzer, M.; Devamani, T.; Jones, B.; Mooney, J.; Zhang, Z.J.; Xu, J.H.; Kazlauskas, R.J.; Dean, A.M. Evolution of a Catalytic Mechanism. Mol. Biol. Evol. 2016, 33, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Devamani, T.; Rauwerdink, A.M.; Lunzer, M.; Jones, B.J.; Mooney, J.L.; Tan, M.A.; Zhang, Z.J.; Xu, J.H.; Dean, A.M.; Kazlauskas, R.J. Catalytic Promiscuity of Ancestral Esterases and Hydroxynitrile Lyases. J. Am. Chem. Soc. 2016, 138, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Rauwerdink, A.; Kazlauskas, R.J. How the Same Core Catalytic Machinery Catalyzes 17 Different Reactions: The Serine-Histidine-Aspartate Catalytic Triad of alpha/beta-Hydrolase Fold Enzymes. ACS Catal. 2015, 5, 6153–6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpigny, J.L.; Jaeger, K.E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef]
- Numoto, N.; Kamiya, N.; Bekker, G.J.; Yamagami, Y.; Inaba, S.; Ishii, K.; Uchiyama, S.; Kawai, F.; Ito, N.; Oda, M. Structural Dynamics of the PET-Degrading Cutinase-like Enzyme from Saccharomonospora viridis AHK190 in Substrate-Bound States Elucidates the Ca(2+)-Driven Catalytic Cycle. Biochemistry 2018, 57, 5289–5300. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, P.; Arora, P.; Chaudhury, A.; Chand, S.; Dilbaghi, N. Characterization of an extracellular alkaline lipase from Pseudomonas mendocina M-37. J. Basic Microbiol. 2010, 50, 420–426. [Google Scholar] [CrossRef]
- Bollinger, A.; Thies, S.; Knieps-Grunhagen, E.; Gertzen, C.; Kobus, S.; Hoppner, A.; Ferrer, M.; Gohlke, H.; Smits, S.H.J.; Jaeger, K.E. A Novel Polyester Hydrolase From the Marine Bacterium Pseudomonas aestusnigri—Structural and Functional Insights. Front. Microbiol. 2020, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, N.E.; Adams, M.C.; Chafin, A.C.; Jones, D.D.; Tsui, C.L.; Gruber, T.D. The highly crystalline PET found in plastic water bottles does not support the growth of the PETase-producing bacterium Ideonella sakaiensis. Environ. Microbiol. Rep. 2020, 12, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Han, X.; Ko, T.P.; Liu, W.; Guo, R.T. Structural studies reveal the molecular mechanism of PETase. FEBS J. 2018, 285, 3717–3723. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, S.; Yamato, S.; Kanaya, E.; Kim, J.J.; Koga, Y.; Takano, K.; Kanaya, S. Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl. Environ. Microbiol. 2012, 78, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.L.; Texier, H.; Gavalda, S.; et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef]
- Boston, M.; Requadt, C.; Danko, S.; Jarnagin, A.; Ashizawa, E.; Wu, S.; Poulose, A.J.; Bott, R. Structure and function engineered Pseudomonas mendocina lipase. Methods Enzymol. 1997, 284, 298–317. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jaroszewski, L.; Iyer, M.; Sedova, M.; Godzik, A. FATCAT 2.0: Towards a better understanding of the structural diversity of proteins. Nucleic Acids Res. 2020, 48, W60–W64. [Google Scholar] [CrossRef] [PubMed]
- Suplatov, D.; Sharapova, Y.; Svedas, V. Mustguseal and Sister Web-Methods: A Practical Guide to Bioinformatic Analysis of Protein Superfamilies. Methods Mol. Biol. 2021, 2231, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Suplatov, D.A.; Kopylov, K.E.; Popova, N.N.; Voevodin, V.V.; Svedas, V.K. Mustguseal: A server for multiple structure-guided sequence alignment of protein families. Bioinformatics 2018, 34, 1583–1585. [Google Scholar] [CrossRef] [Green Version]
- Dietmann, S.; Park, J.; Notredame, C.; Heger, A.; Lappe, M.; Holm, L. A fully automatic evolutionary classification of protein folds: Dali Domain Dictionary version 3. Nucleic Acids Res. 2001, 29, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suplatov, D.; Sharapova, Y.; Timonina, D.; Kopylov, K.; Svedas, V. The visualCMAT: A web-server to select and interpret correlated mutations/co-evolving residues in protein families. J. Bioinform. Comput. Biol. 2018, 16, 1840005. [Google Scholar] [CrossRef]
- Kuhn, B.; Tichy, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.; Giroud, M.; Schirmeister, T.; Abel, R.; et al. Prospective Evaluation of Free Energy Calculations for the Prioritization of Cathepsin L Inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Grimm, M.; Dai, W.T.; Hou, M.C.; Xiao, Z.X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Procter, J.B.; Carstairs, G.M.; Soares, B.; Mourao, K.; Ofoegbu, T.C.; Barton, D.; Lui, L.; Menard, A.; Sherstnev, N.; Roldan-Martinez, D.; et al. Alignment of Biological Sequences with Jalview. Methods Mol. Biol. 2021, 2231, 203–224. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Chan, H.C.S.; Filipek, S.; Vogel, H. PyMOL and Inkscape Bridge the Data and the Data Visualization. Structure 2016, 24, 2041–2042. [Google Scholar] [CrossRef] [Green Version]
- Tomasello, G.; Armenia, I.; Molla, G. The Protein Imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef]
- Unni, S.; Huang, Y.; Hanson, R.M.; Tobias, M.; Krishnan, S.; Li, W.W.; Nielsen, J.E.; Baker, N.A. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 2011, 32, 1488–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, S.; Richter, S.; Wade, R.C. On the use of PIPSA to guide target-selective drug design. ChemMedChem 2008, 3, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wenzel, A.; Stein, M.; Gabdoulline, R.R.; Wade, R.C. webPIPSA: A web server for the comparison of protein interaction properties. Nucleic Acids Res. 2008, 36, W276–W280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, G.J.; Reisky, L.; Bottcher, D.; Muller, H.; Michels, E.A.P.; Walczak, M.C.; Berndt, L.; Weiss, M.S.; Bornscheuer, U.T.; Weber, G. Structure of the plastic-degrading Ideonella sakaiensis MHETase bound to a substrate. Nat. Commun. 2019, 10, 1717. [Google Scholar] [CrossRef]
- Daniels, N.M.; Nadimpalli, S.; Cowen, L.J. Formatt: Correcting protein multiple structural alignments by incorporating sequence alignment. BMC Bioinform. 2012, 13, 259. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, M.; Chomilier, J. Protein multiple alignments: Sequence-based versus structure-based programs. Bioinformatics 2019, 35, 3970–3980. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Roy, U.; Chowdhury, A.R.; Bhaduri, A.P.; Roy, P.K. Purification and characterization of an alkaline lipase from a newly isolated Pseudomonas mendocina PK-12CS and chemoselective hydrolysis of fatty acid ester. Bioorg Med. Chem. 2003, 11, 1041–1046. [Google Scholar] [CrossRef]
- Miyakawa, T.; Mizushima, H.; Ohtsuka, J.; Oda, M.; Kawai, F.; Tanokura, M. Structural basis for the Ca(2+)-enhanced thermostability and activity of PET-degrading cutinase-like enzyme from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2015, 99, 4297–4307. [Google Scholar] [CrossRef] [PubMed]
- Nathan, V.K.; Rani, M.E. A cleaner process of deinking waste paper pulp using Pseudomonas mendocina ED9 lipase supplemented enzyme cocktail. Environ. Sci. Pollut Res. Int. 2020, 27, 36498–36509. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Juan, D.; Valencia, A.; Pazos, F. Detection of significant protein coevolution. Bioinformatics 2015, 31, 2166–2173. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; He, L.; Wang, L.; Li, T.; Li, C.; Liu, H.; Luo, Y.; Bao, R. Protein Crystallography and Site-Direct Mutagenesis Analysis of the Poly(ethylene terephthalate) Hydrolase PETase from Ideonella sakaiensis. Chembiochem 2018, 19, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.; Cho, I.J.; Seo, H.; Son, H.F.; Sagong, H.Y.; Shin, T.J.; Choi, S.Y.; Lee, S.Y.; Kim, K.J. Structural insight into molecular mechanism of poly(ethylene terephthalate) degradation. Nat. Commun. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecker, T.; Galaz-Davison, P.; Engelberger, F.; Narui, Y.; Sotomayor, M.; Parra, L.P.; Ramirez-Sarmiento, C.A. Active Site Flexibility as a Hallmark for Efficient PET Degradation by I. sakaiensis PETase. Biophys. J. 2018, 114, 1302–1312. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Hiraga, K.; Takehana, T.; Taniguchi, I.; Yamaji, H.; Maeda, Y.; Toyohara, K.; Miyamoto, K.; Kimura, Y.; Oda, K. A bacterium that degrades and assimilates poly(ethylene-terephthalate). Science 2016, 351, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.-J.; Schrader, H.; Profe, J.; Dresler, K.; Deckwer, W.-D. Enzymatic Degradation of Poly (ethylene terephthalate): Rapid Hydrolyse using a Hydrolase from T. fusca. Macromol. Rapid Commun. 2005, 26, 1400–1405. [Google Scholar] [CrossRef]
- Kleeberg, I.; Welzel, K.; Vandenheuvel, J.; Muller, R.J.; Deckwer, W.D. Characterization of a new extracellular hydrolase from Thermobifida fusca degrading aliphatic-aromatic copolyesters. Biomacromolecules 2005, 6, 262–270. [Google Scholar] [CrossRef]
- Liebminger, S.; Eberl, A.; Sousa, F.; Heumann, S.; Fischer-Colbrie, G.; Cavaco-Paulo, A.; Guebitz, G.M. Hydrolysis of PET and bis-(benzoyloxyethyl) terephthalate with a new polyesterase fromPenicillium citrinum. Biocatal. Biotransform. 2009, 25, 171–177. [Google Scholar] [CrossRef]
- Maester, T.C.; Pereira, M.R.; Malaman, A.M.G.; Borges, J.P.; Pereira, P.A.M.; Lemos, E.G.M. Exploring Metagenomic Enzymes: A Novel Esterase Useful for Short-Chain Ester Synthesis. Catalysts 2020, 10, 1100. [Google Scholar] [CrossRef]
- Nikolaivits, E.; Kanelli, M.; Dimarogona, M.; Topakas, E. A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases. Catalysts 2018, 8, 612. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.; Edwards, S.; Vague, M.; Leon-Zayas, R.; Scheffer, H.; Chan, G.; Swartz, N.A.; Mellies, J.L. Environmental Consortium Containing Pseudomonas and Bacillus Species Synergistically Degrades Polyethylene Terephthalate Plastic. mSphere 2020, 5. [Google Scholar] [CrossRef]
- Yeang, C.H.; Haussler, D. Detecting coevolution in and among protein domains. PLoS Comput. Biol. 2007, 3, e211. [Google Scholar] [CrossRef] [PubMed]
- Salinas, V.H.; Ranganathan, R. Coevolution-based inference of amino acid interactions underlying protein function. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Little, D.Y.; Chen, L. Identification of coevolving residues and coevolution potentials emphasizing structure, bond formation and catalytic coordination in protein evolution. PLoS ONE 2009, 4, e4762. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.Y.; Patel, P.; Kim, P.M.; Engelman, D.M.; McDermott, D.; Gerstein, M. An integrated system for studying residue coevolution in proteins. Bioinformatics 2008, 24, 290–292. [Google Scholar] [CrossRef] [Green Version]
- Son, H.F.; Joo, S.; Seo, H.; Sagong, H.Y.; Lee, S.H.; Hong, H.; Kim, K.J. Structural bioinformatics-based protein engineering of thermo-stable PETase from Ideonella sakaiensis. Enzym. Microb. Technol. 2020, 141, 109656. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, Y.; Liu, X.; Dong, S.; Tian, Y.e.; Qiao, Y.; Mitra, R.; Han, J.; Li, C.; Han, X.; et al. Computational Redesign of a PETase for Plastic Biodegradation under Ambient Condition by the GRAPE Strategy. ACS Catal. 2021, 11, 1340–1350. [Google Scholar] [CrossRef]
- Puspitasari, N.; Tsai, S.L.; Lee, C.K. Fungal Hydrophobin RolA Enhanced PETase Hydrolysis of Polyethylene Terephthalate. Appl. Biochem. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitão, A.L.; Enguita, F.J. Structural Insights into Carboxylic Polyester-Degrading Enzymes and Their Functional Depolymerizing Neighbors. Int. J. Mol. Sci. 2021, 22, 2332. https://doi.org/10.3390/ijms22052332
Leitão AL, Enguita FJ. Structural Insights into Carboxylic Polyester-Degrading Enzymes and Their Functional Depolymerizing Neighbors. International Journal of Molecular Sciences. 2021; 22(5):2332. https://doi.org/10.3390/ijms22052332
Chicago/Turabian StyleLeitão, Ana Lúcia, and Francisco J. Enguita. 2021. "Structural Insights into Carboxylic Polyester-Degrading Enzymes and Their Functional Depolymerizing Neighbors" International Journal of Molecular Sciences 22, no. 5: 2332. https://doi.org/10.3390/ijms22052332