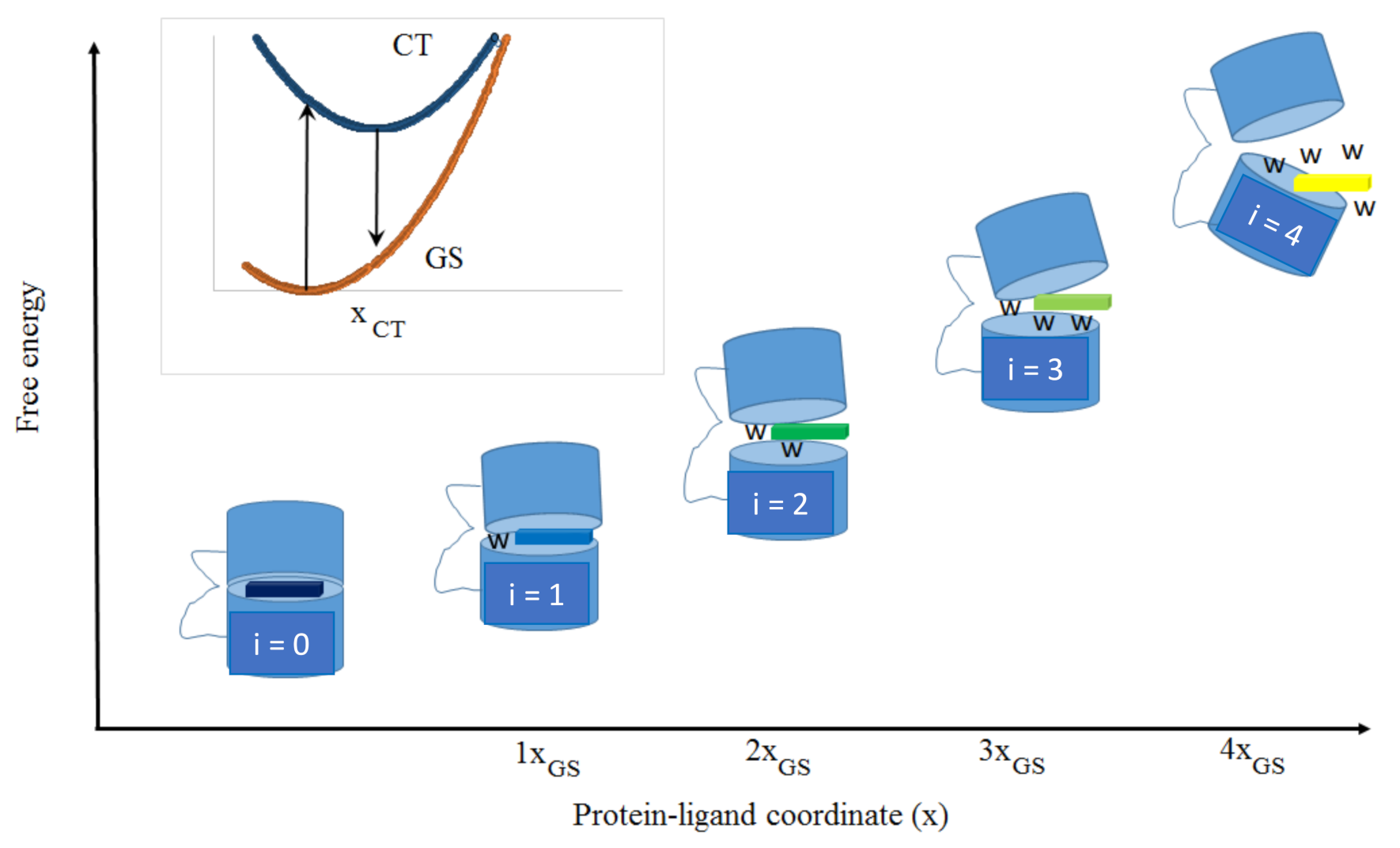
3.1. Simulations of REES Curves for Different Protein–Ligand Free Energy Landscapes
To understand how the theoretical REES curve depends on parameters of the protein–ligand free energy landscape, we have carried out simulations for a defined system consisting of 10 ligand–protein microstates. The spectroscopic parameters of the ligand L were set to (xCT = 0.89 (eV)0.5, κ = 1, ∆GCTGS0 = 3.346 eV, ∆GCTGSN = 3.223 eV), which corresponds to an emission peak in the range of 420nm to 438nm (note that these values are somewhat arbitrary).
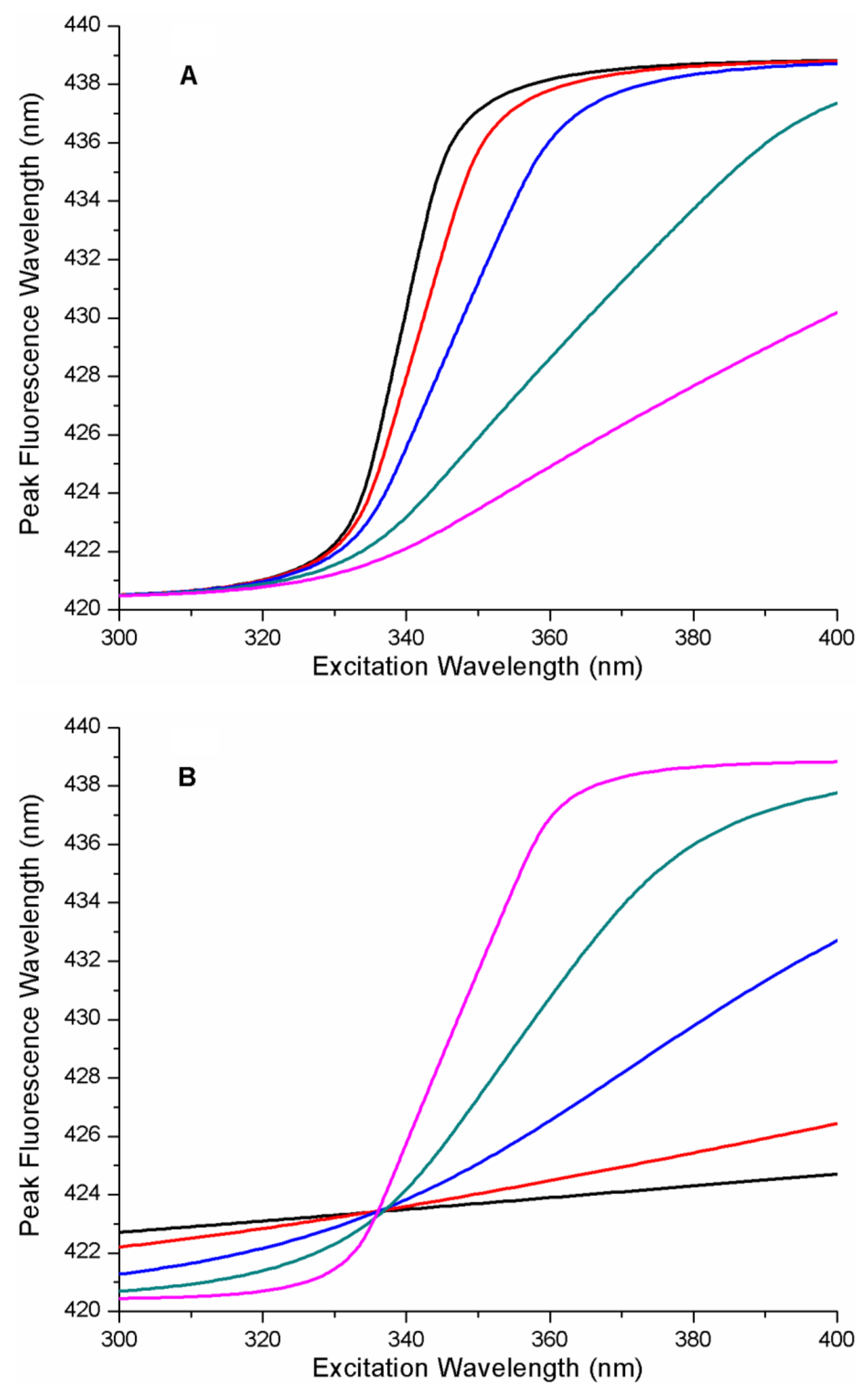
Figure 2 contains a series of REES curves as a function of the energy parameters of the protein–ligand free energy landscape. It is notable that these curves are sigmoidal functions with emission wavelength limits matching those input into the model, as expected. In
Figure 2A,
xGS was fixed and b was set to 1 (b = 1 (linear ramp) and A was variable. This defines the FEL as a linear ramp function. As can be seen in
Figure 2A, increasing the value of the energy parameter A (i.e., the energy gradient), causes a significant change in the position of the mid-point of the sigmodal REES function along the excitation wavelength axis (i.e., to longer wavelengths). In
Figure 2B, A and b were fixed, and the influence on the spacing of the free energy surfaces of the microstates along coordinate
x (i.e., the parameter
xGS) was determined. Increasing the value of
xGS changed the shape of the REES curve, causing the gradient near the mid-point to increase significantly.
In the linear ramp model for the free energy landscape (b = 1), the free energy difference between the successive minima of the microstate free energy surfaces is given by Equation (17),
And the ground-state reorganization energy, RE (Equation (18)) is
From Marcus theory, the activation energy between microstates is (in the linear ramp model) is given by Equation (19),
In the context of a linear protein–ligand FEL, shifts in the mid-point of the REES curve indicate changes in the relative thermodynamic stability of the most populated state relative to the least populated state. Changes in the steepness of the REES transition (all other things being equal) would indicate a change in the roughness of the FEL (i.e., increase or decrease in activation energy between successive microstates). A rougher FEL produces a steeper REES transition.
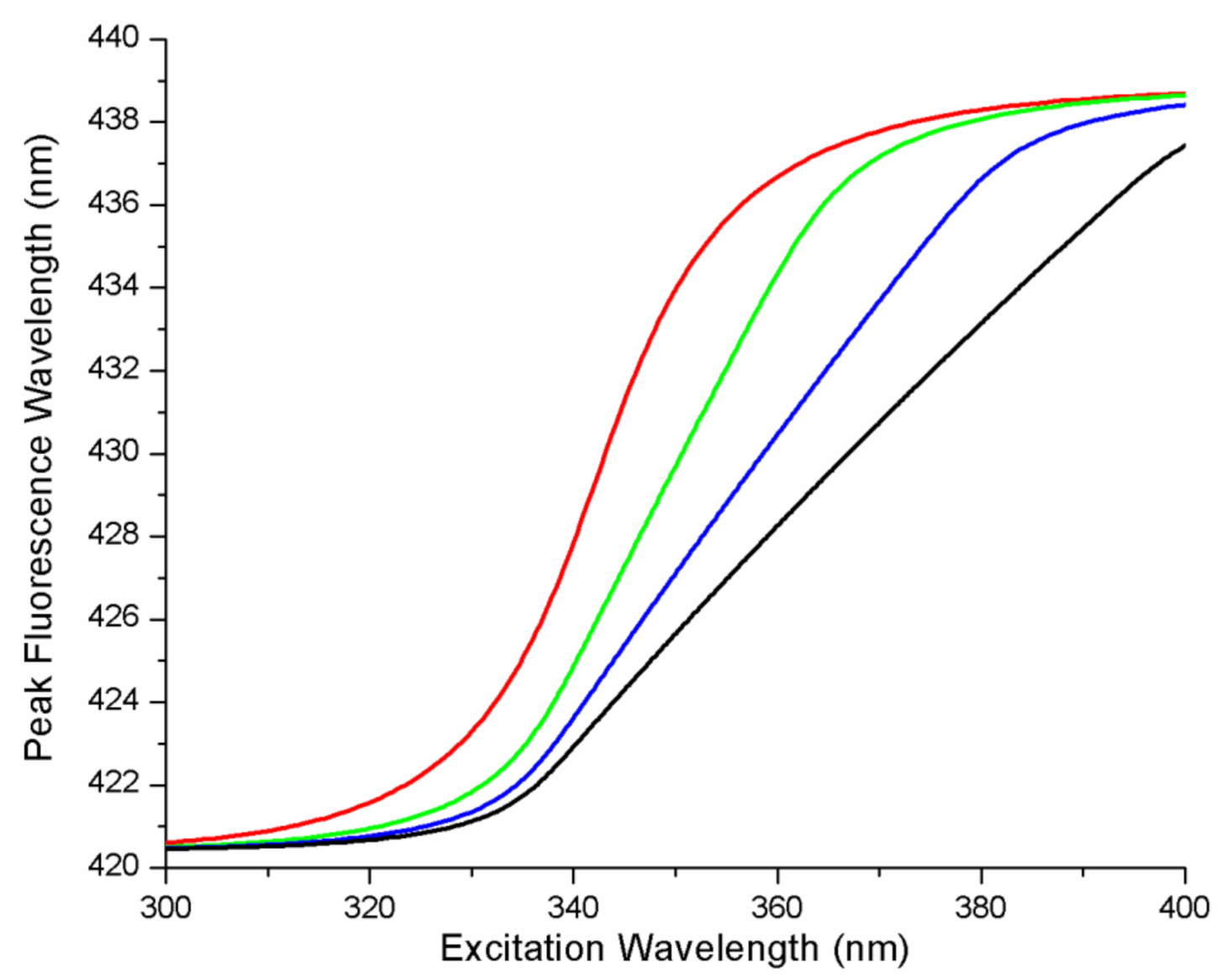
We also considered more complex protein–ligand energy landscapes such as quadratic (or harmonic) free energy as a function of the microstate. The results of a simulation on the influence of changing the A parameter on the REES curves are shown in
Figure 3 for the quadratic FEL (i.e., the ith microstate free energy proportional to i
2). The simulations reveal that increasing A (which is analogous to a Hook’s law spring constant) causes both a shift in the mid-point of the REES curve (along the excitation wavelength axis to longer wavelengths) and a consequential change in the steepness of the REES transition (more shallow appearing transition). In
Figure 3B, A and b were fixed, and the influence on the spacing of the free energy surfaces of the microstates along coordinate x (i.e., the parameter
xGS) was determined. Increasing the value of
xGS, changes the shape of the REES curve, causing the gradient near the mid-point to increase significantly.
If we examine the free energy landscape for the quadratic model (b = 2), we note that the free energy difference between the successive minima of the microstate free energy surfaces is (Equation (20)),
From Marcus theory, the activation energy between successive microstates is (in the quadratic FEL model) given by Equation (21),
By inspection of Equation (20), we see that in the quadratic FEL model, the successive free energy differences between the microstate minimum increase with the ith microstate, and consequently the Marcus activation energy between microstates is also no longer independent of the microstate (Equation (21)). According to the quadratic FEL model (Equation (21)), the activation barrier between successive microstates increases with increasing i, while the activation barriers decrease in the opposite direction.
To explore the effect of different numbers of microstates on the REES curves, we fixed all the parameters in the protein–ligand FEL and ligand L (parameters were b = 2, A = 0.0025,
xGS = 0.15)) and varied N from 5, 10, 15, and 20 microstates. The influence of the number of microstates, N, on the REES curves is depicted in
Figure 4. An increase in the number of microstates, N, is accompanied by a substantial change in shape of the REES curves. In general, both the position of the mid-point and the slope of the curve at the mid-point changed with N. Increasing N decreased the slope near the transition mid-point and increased the excitation wavelength at which the mid-point transition occurred.
The parameters chosen in these simulations were in the range of what we might expect for intermediate states in protein–ligand complexes. The free energy difference between the most stable state in the protein–ligand complex and the least stable bound state in the protein ligand complex was 10A (for N = 10 and for the linear model, A = 0.0025–0.05 eV). This corresponds to a free energy value range from 0.025 to 0.5 eV or 2.5–50 KJ/mol or about 0.6–12 kcal/mol (2 kbT to 20 kbT (kb is Boltzmann constant)). By way of comparison, the total free energy between the lowest energy bound state and a totally-free ligand state between 6.9 kbT and 20.7 kbT corresponds to an equilibrium dissociation constant of between 1 mM and 1 nM (i.e., the range of equilibrium constants for the majority of protein–ligand interactions).
3.2. Application to REES Data from Kinase Inhibitor–Kinase Complexes
To provide a concrete example of the application of the theoretical models to real data, we refer to recent published data from the anilino-quinazoline small molecule kinase inhibitor (AG1478) in complex with a kinase (MAPK14 or p38α kinase) [
17]. AG1478 is an intrinsically-fluorescent kinase inhibitor and exhibits environmentally-sensitive fluorescence with a Stokes shift as large as 100 nm in highly polar solvents [
18]. Therefore, AG1478 is a suitable ligand for REES studies. Spectroscopic studies on the AG1478-p38α kinase complex revealed a progressive red-shift in the emission spectrum with increasing excitation wavelength, consistent with a REES-type effect. The magnitude of the total red-edge shift was also largely independent of temperature (in the range 283–313 K) [
17], which is an indication [
19] that the motions of the environment are restricted on the nanosecond time-scale. In this case, we can apply the theory outlined here to analyze the REES data.
Figure 5 depicts the REES curve as a solid line represented by the Boltzmann sigmoid fitting function used in the publication [
17]. Individual datapoints corresponding to the REES data are indicated by the open circles in the inset to
Figure 5. To fit this data to our thermodynamic model, we first optimized the photo-physical parameters of the AG1478 ligand in our model to provide a good fit to the extremes of the REES curve (i.e., to match the initial and final emission values in the REES curve). In our analysis, we then fixed the values for the ligand photo-physics (see
Table 1 for parameter values). We then chose the form of the FEL function (linear or quadratic or cubic) and then performed a least-squares fit to the experimental curve by adjusting two parameters, the energy parameter A and the microstate spacing parameter
xGS. The number of microstates was initially fixed to N = 10 per simulations in the previous section.
Table 2 outlines the results of each analysis, together with the sum of squares for the fit. Plots of the simulated REES curves are shown in the inset of
Figure 5 to afford visual comparison. The linear model provided a reasonable fit with a SLS value of 53.5. The cubic model also provided a reasonable fit to the data with a SLS value of 204. However, the quadratic model outperformed both models with a SLS value of 3.5 (i.e., by a factor of 10 improvement). All models had the same number of adjustable parameters; therefore, we rejected the linear and cubic models, in favor of the quadratic model.
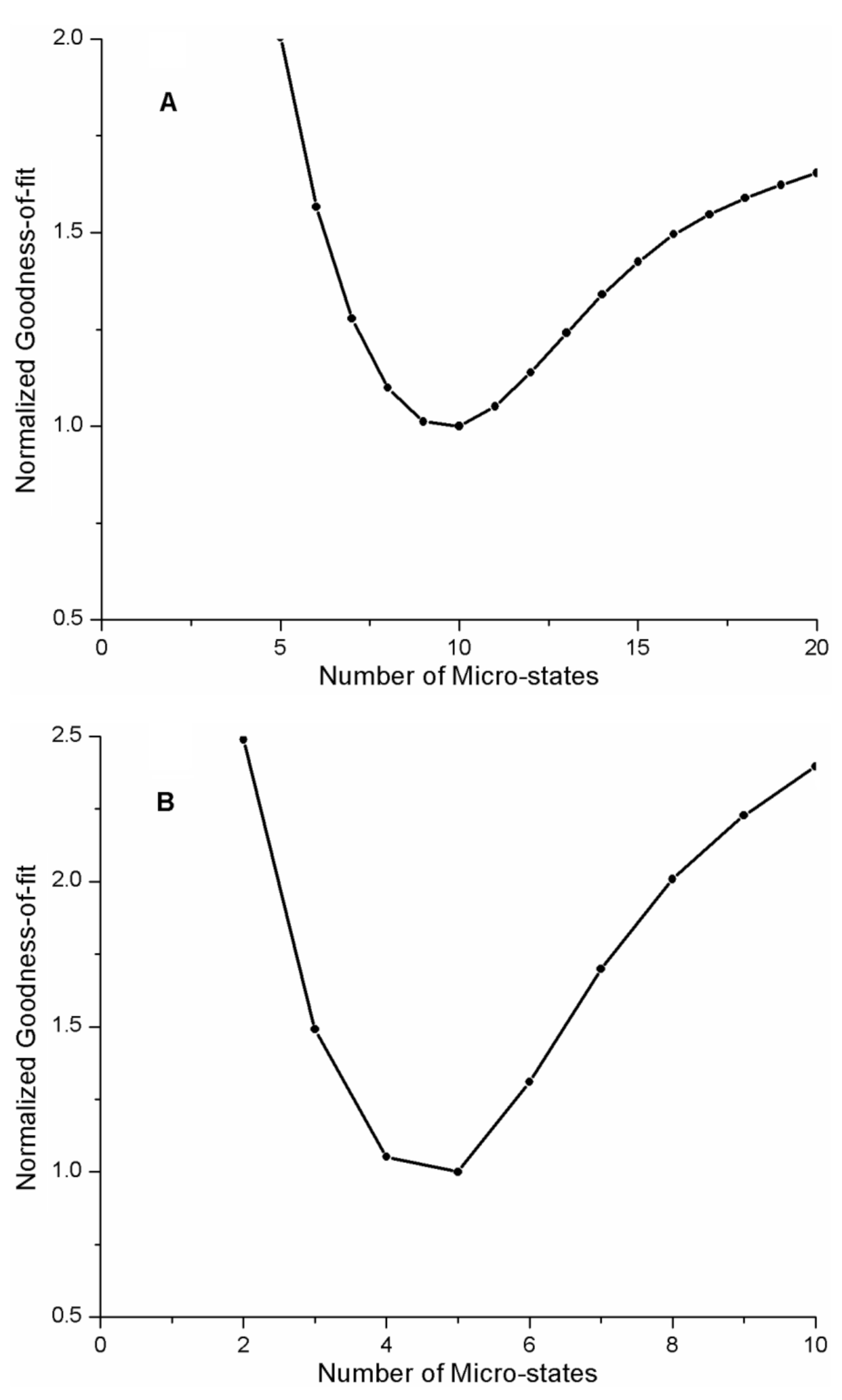
In fitting the REES curve for the AG1478-p38α kinase complex, we initially assumed that the number of microstates was N = 10. We repeated the analysis of the REES curve for different values of N for the quadratic FEL model as described above, (range of N = 3 to 20). The goodness-of-fit parameter as a function of the number of microstates is depicted in
Figure 6A. As can be seen in
Figure 6A, the goodness-of-fit is dependent on the number of microstates in the model, with N = 10 providing the best fit to the data.
An important concern is whether by pre-selecting a quadratic FEL, we are biasing the fitting in some way. Therefore, we repeated the analysis of the p38α-kinase-AG1478 REES data, allowing all the parameters of the FEL to vary. The goodness-of-fit parameter versus number of microstates revealed a clear minimum (near N = 9–10) with the FEL exponent close to 2 (b = 1.94). To examine the robustness of our fitting procedures and model in more detail, we simulated REES data, fit the simulated REES curve to a sigmoidal function (as we do for experimental data), then fit the resulting sigmoid to a FEL model.
Table 3 compares the input parameters with output parameters for FELs over an energy range of 0.04–0.24 eV. It is noted that the output parameters were within 15% of the input parameters.
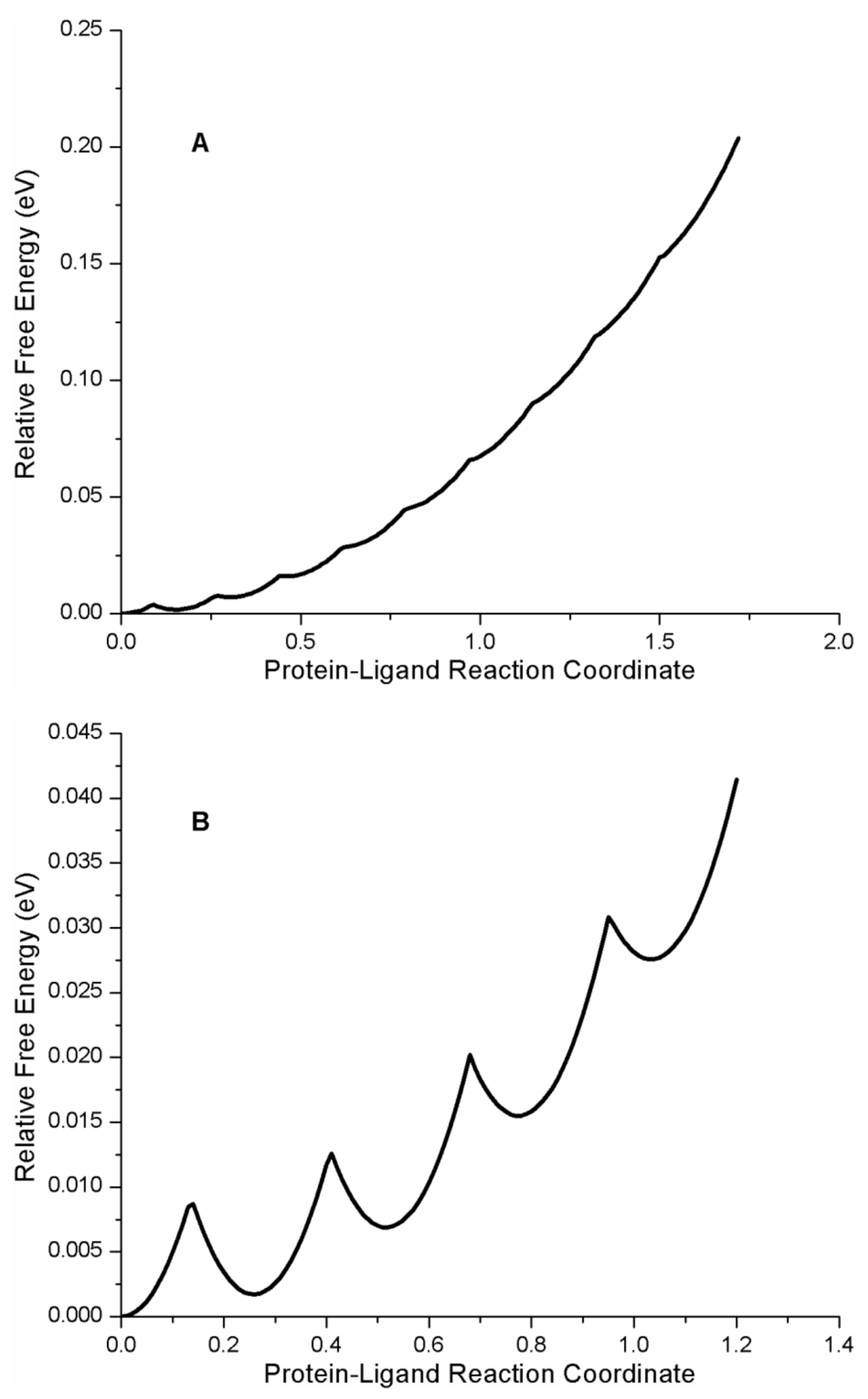
A plot of the free energy landscape of the protein–ligand complex AG1478-p38α kinase is shown in
Figure 7A, depicting the relative free energies of the 10 protein–ligand microstates as a function of the generalized protein–ligand coordinate. According to the quadratic FEL model and the fit parameters, the free energy difference between the most stable bound state and the least stable bound state in the AG1478-p38α kinase complex was −0.144 eV (or −5.6
kbT or −14.4 kJ/mol or −3.5 kcal/mol). The reported inhibition constant for the AG1478-p38α kinase complex was 0.560 µM (
20). A 0.6 µM equilibrium dissociation constant corresponded to a free energy difference between bound and free AG1478 of −14.4
kbT. According to our analysis, the intermediate states of AG1478-p38α kinase could contribute as much as ~40% (= 5.6/14.4) to the total free energy of binding.
To test this idea further, we also analyzed the published REES data from AG1478 bound to another kinase, aminoglycophosphokinase (APH) [
17] which is a threonine-serine kinase derived from bacteria, but has a similar fold to eukaryotic protein kinases. A plot of the REES curve for the AG1478-APH complex is depicted in
Figure 5B, REES data in the
Figure 5B inset, photophysical parameters in
Table 4, and analysis of different models collected in
Table 5. We found the best fit was to a quadratic model (
Table 4) for the FEL surface with five microstates (i.e., N = 5,
Figure 6B). From the model fit, the free energy difference between the most stable state and the least stable state in the AG1478-APH kinase complex was −0.03 eV or approximately −1
kbT.
A comparison of the free energy landscapes for the two kinase-AG1478 complexes is shown in
Figure 7 (generated using Equation (7)). It is notable that as a function of the protein–ligand reaction coordinate (x in Equations (1)–(14)), the p38α-AG1478 free energy profile was steeper while the APH-AG1478 the free energy profile was shallower. From a physics analogy of a Harmonic spring, one would deduce that it is harder (i.e., costs more energy) to displace AG1478 from its most stable bound state in the p38α kinase complex along the reaction coordinate (to its least stable bound state) than to displace the most-stable bound state in the corresponding APH-AG1478 complex because the AG1478-p38α kinase complex interaction has a larger effective spring constant. This qualitatively agrees with the relative inhibition constants of the two proteins. The inhibition dissociation constant for p38α kinase-AG1478 was K
i(p38α kinase) = 0.560 µM and represents “tighter” binding than for APH-AG1478 interaction, with an inhibition dissociation constant of K
i(APH) = 15.6 µM (
21).
At equilibrium, the total binding free energy for the protein–ligand interaction (∆Gprotein–ligand) is the sum of the free energy for ligand association with protein (∆Gassoc) to a surface state (or encounter complex) and a free energy for ligand intrusion (∆Gintrusion) from a surface-bound state to the interior-bound state. If we equate (∆Gintrusion) with the free energy difference between the most-stable bound state and the least-stable bound state, then this quantity may be extracted from the protein–ligand FELs derived from REES data. From the FELs for the AG1478 complexes with APH and p38α kinase, we estimated (∆Gintrusion)APH-AG1478 = −0.7 kcal/mol and (∆Gintrusion)p38α-G1478 = −3.3 kcal/mol. The total free energies for binding (from the inhibition constants) were (∆Gprotein-ligand)APH-AG1478 = −6.5 kcal/mol for the APH-AG1478 interaction and (∆Gprotein–ligand)p38 α−AG1478 = −8.3 kcal/mol for the p38α-AG1478 interaction. From ∆Gintrusion and ∆Gprotein–ligand, we estimated (∆Gassoc)APH-AG1478 = −5.8 kcal/mol and (∆Gassoc)p38a-AG1478 = −5.0 kcal/mol. Comparing the differences between the two proteins, we can see that the difference in total free energies for binding ((δ∆Gprotein-ligand)p38a/APH = 1.84 kcal/mol) was more than accounted for by the difference in the free energies for intrusion ((δ∆Gintrusion)p38a/APH = 2.6 kcal/mol). In contrast, the difference in free energies for surface association ((δ∆Gassoc)p38a/APH = −0.8 kcal/mol) makes a comparatively smaller contribution. Thus, the energetics of the ligand-bound states of the protein–ligand complexes is a differentiating factor in explaining the different inhibition constants of AG1478 for two different kinases.
The protein–ligand free energy landscapes, derived from the REES data, also provide estimates of activation barriers for transitions between the different ligand–protein microstates (along the reaction coordinate, x). As eluded to above, for the quadratic FEL, the activation barrier is related to the free energy difference and the reorganization energy between microstates (see Equation 20). For the APH-AG1478 complex, the largest activation barrier between the 4th and 5th microstate was 0.016 eV. For the p38α kinase-AG1478 complex, the largest activation barrier between successive microstates was between the 9th and 10th microstates and was 0.031 eV. Thus for both complexes, the successive barrier heights were not significantly greater than the thermal energy (i.e.,
kbT = 0.025 eV at 298K). From the model, one would also predict that the total magnitude of the REES effect is not very dependent on temperature, which agrees qualitatively with published observations over the temperature range 283–308 K [
17].
Our focus thus far has been on the properties of the FEL (i.e., in the electronic ground states of the ligand in the microstates defined by the protein–ligand interaction). The extrema of the REES curves and associated analysis provide useful information on the CT states, and by inference, on the types of environments encountered by the ligand in the protein microstates (i.e., polarity of the amino acids and/or relative hydration). The relative displacement of the CT state relative to the GS free energy surfaces,
xCT, indicates the change in structure of the ligand (and possibly environment) between the GS and CT states. From this displacement
xCT, we can determine the Marcus reorganization energy for the CT to GS transition, equivalent to half the Stokes shift. Comparing the
xCT values for the two protein–ligand complexes (c/f
Table 1 and
Table 3), the displacement associated with the CT–GS transition in AG1478-APH (
xCT = 1.1) was slightly larger than the corresponding quantity in the AG1478-p38α complex (
xCT = 0.9). Consequentially, the reorganization energy for the CT-GS transition in AG1478-APH was 0.6 eV and for AG1478-p38a, it was 0.4 eV. The reorganization energy consists of two parts, the inner-sphere contribution (from changes in the geometry of the AG1478 and tightly-associated molecules), and an outer-sphere part, due to changes in dipolar reorganization of the environment around the AG1478. The inner-sphere term can be determined from the fluorescence of AG1478 in a non-polar solvent (where the outer-sphere contribution due to solvent dipolar reorganization is zero). From the published emission of AG1478 in non-polar solvents (toluene and dioxane), we estimated the inner-sphere reorganization energy in AG1478 to be 0.4 eV [
18]. Thus, in the AG1478–p38α kinase complex and the AG1478–APH complex, the inner-sphere reorganization of the ligand is the dominant contribution to the reorganization energy. This suggests that the dipolar reorganization of the protein environment during the excited state lifetime is restricted in the AG1478-APH complex and absent (or undetected) in the AG1478-p38α kinase complexes. The free energy gap between CT and GS provide valuable information on the stabilization of the CT state relative to the GS by the protein environment and water. In the AG1478–APH complex, the CT states were more stabilized with respect to the GS (
∆GCTGS0 = 3.18 eV,
∆GCTGSN = 3.02 eV) than the corresponding AG1478–p38α kinase complex (
∆GCTGS0 = 3.35 eV,
∆GCTGSN = 3.22 eV). These observations suggest that AG1478 experiences a more polar environment along its microstate trajectory in the APH protein compared to the p38α kinase protein. It is tempting to speculate that the higher affinity of AG1478 for p38α than APH is due to the more hydrophobic binding environments in p38α, perhaps contributed in part by the nature and number of water molecules and the amino acids comprising the ligand binding pocket. However, the polarity gradients along the microstate trajectories (
∆GCTGSN −
∆GCTGS0 = −0.16eV (APH);
∆GCTGSN −
∆GCTGS0 = −0.13eV (p38α kinase)) were similar in direction and magnitude in both proteins. This may imply that least populated ligand states are more hydrated than the most populated ligand state, a view that is congruent with the idea that ligands become more hydrated as they leave the protein.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}