
Overexpression of Human ABCB1 and ABCG2 Reduces the Susceptibility of Cancer Cells to the Histone Deacetylase 6-Specific Inhibitor Citarinostat

 , and
, and
Abstract
:1. Introduction
2. Results
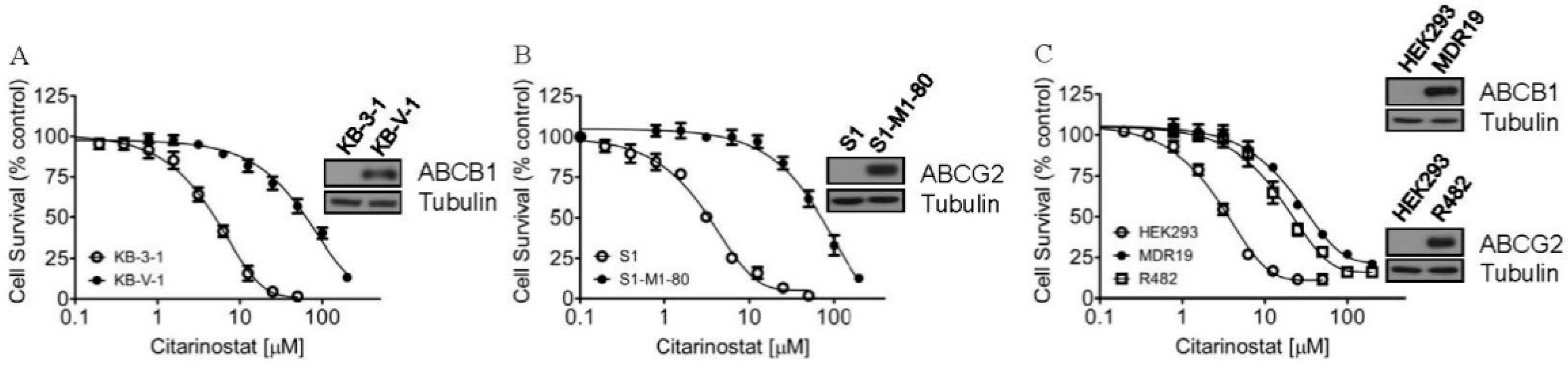
2.1. Citarinostat Is Less Cytotoxic in Cells Overexpressing ABCB1 or ABCG2
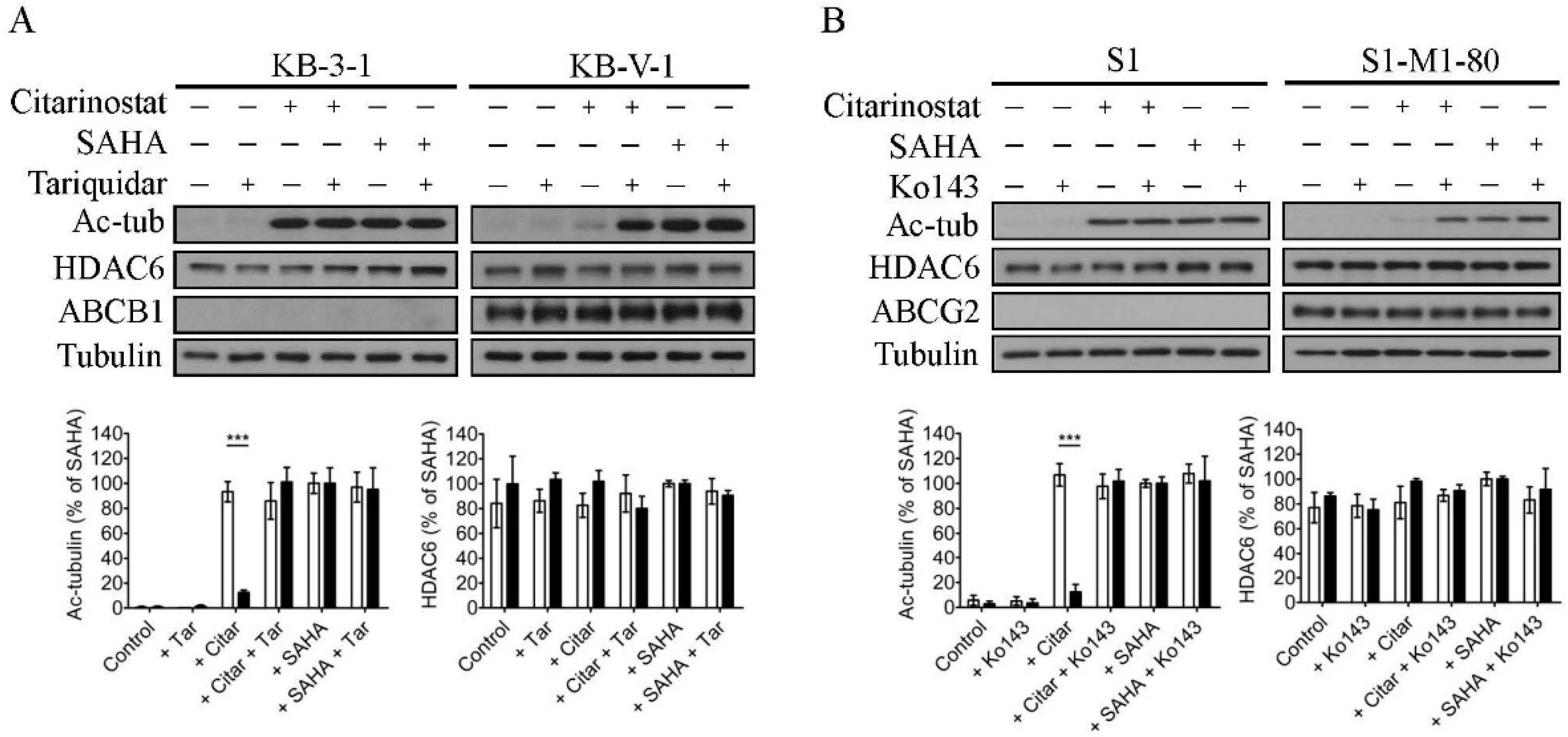
2.2. The Effect of Citarinostat on the Activity of HDAC6 Is Reduced by ABCB1 and ABCG2 in Human Cancer Cell Lines
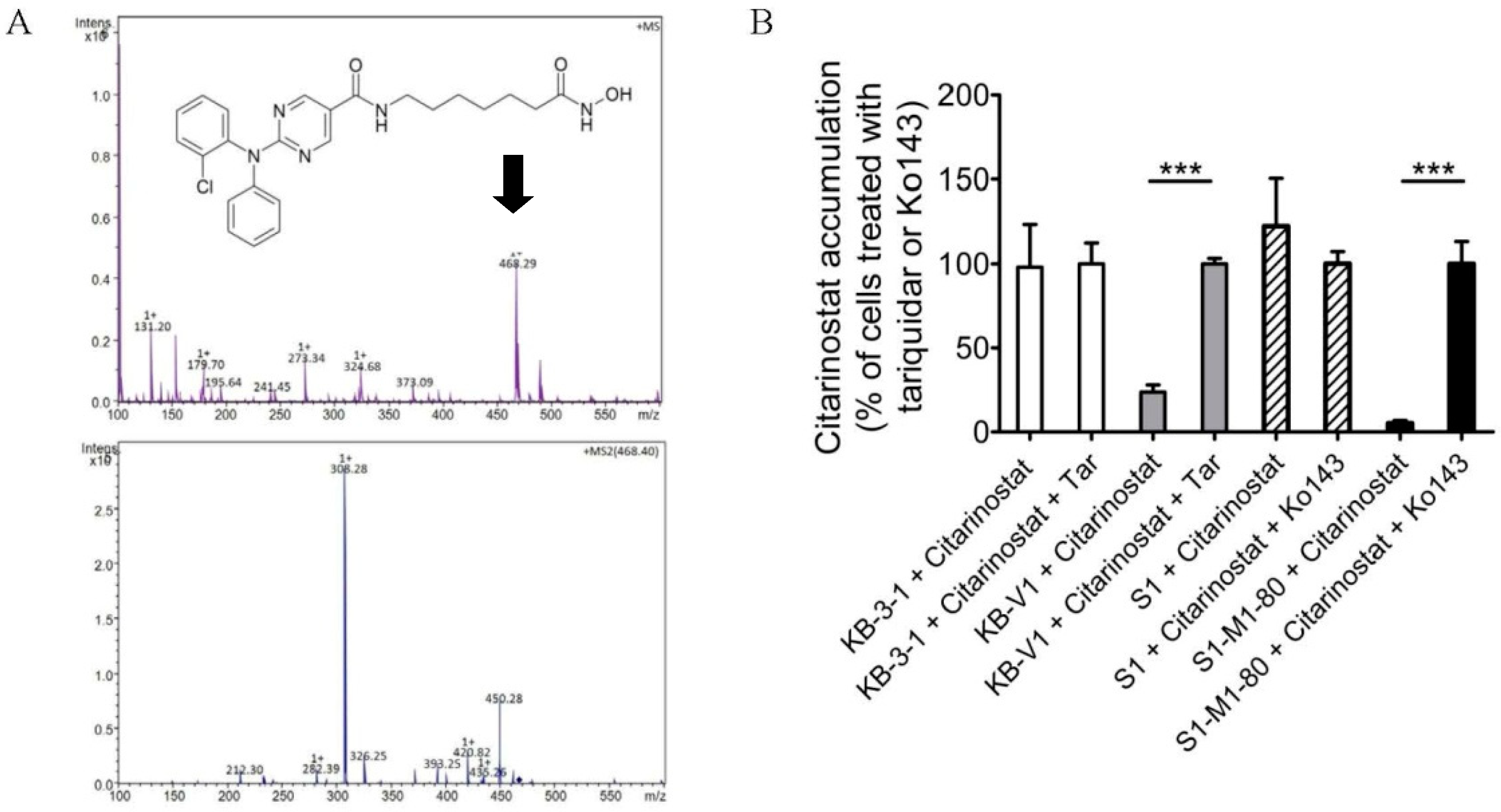
2.3. The Intracellular Accumulation of Citarinostat Is Reduced by ABCB1 and ABCG2 in Human Cancer Cell Lines
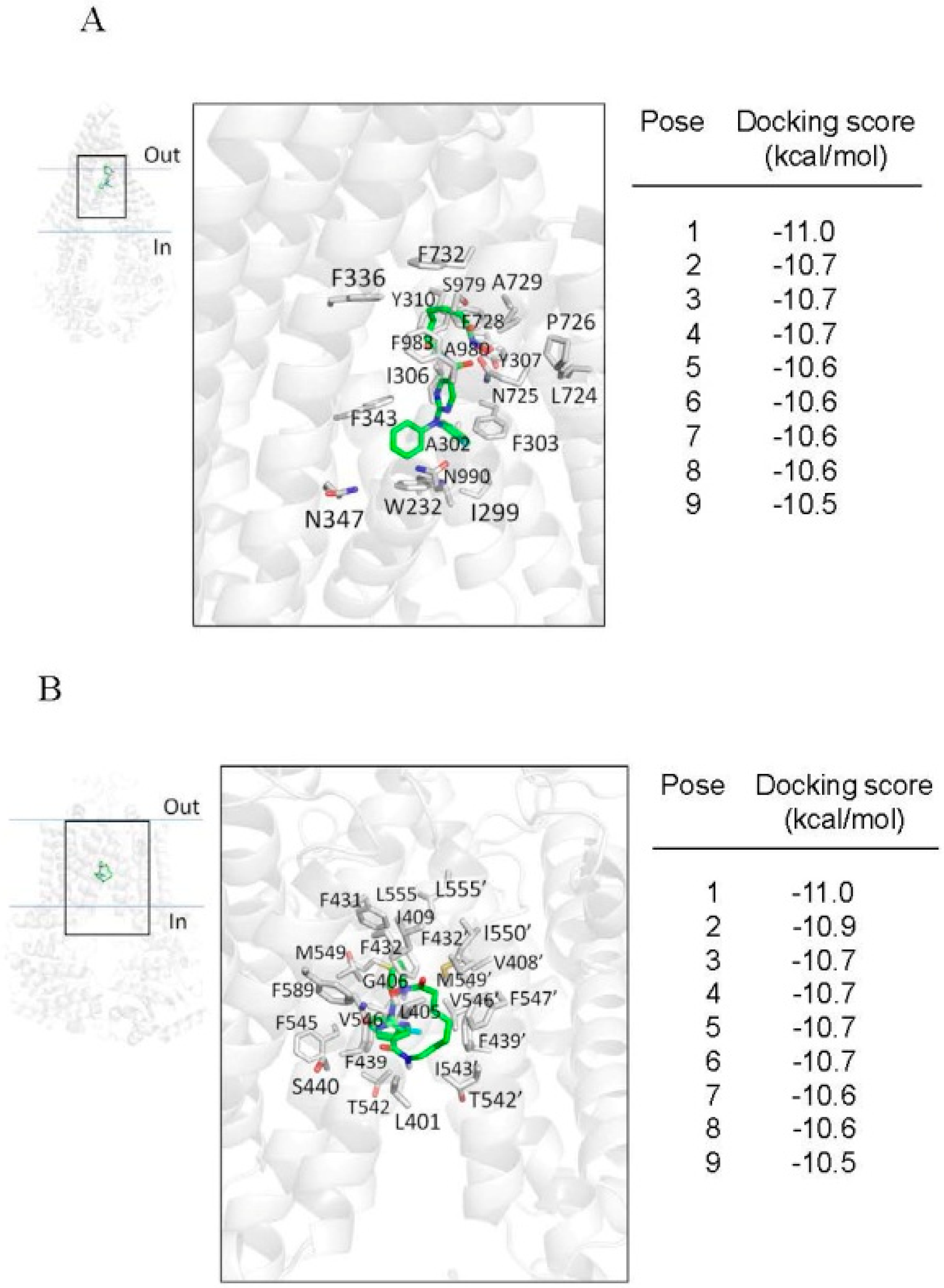
2.4. Docking of Citarinostat in the Drug-Binding Pocket of ABCB1 and ABCG2
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Culture Conditions
4.3. Cytotoxicity Assays
4.4. Immunoblot
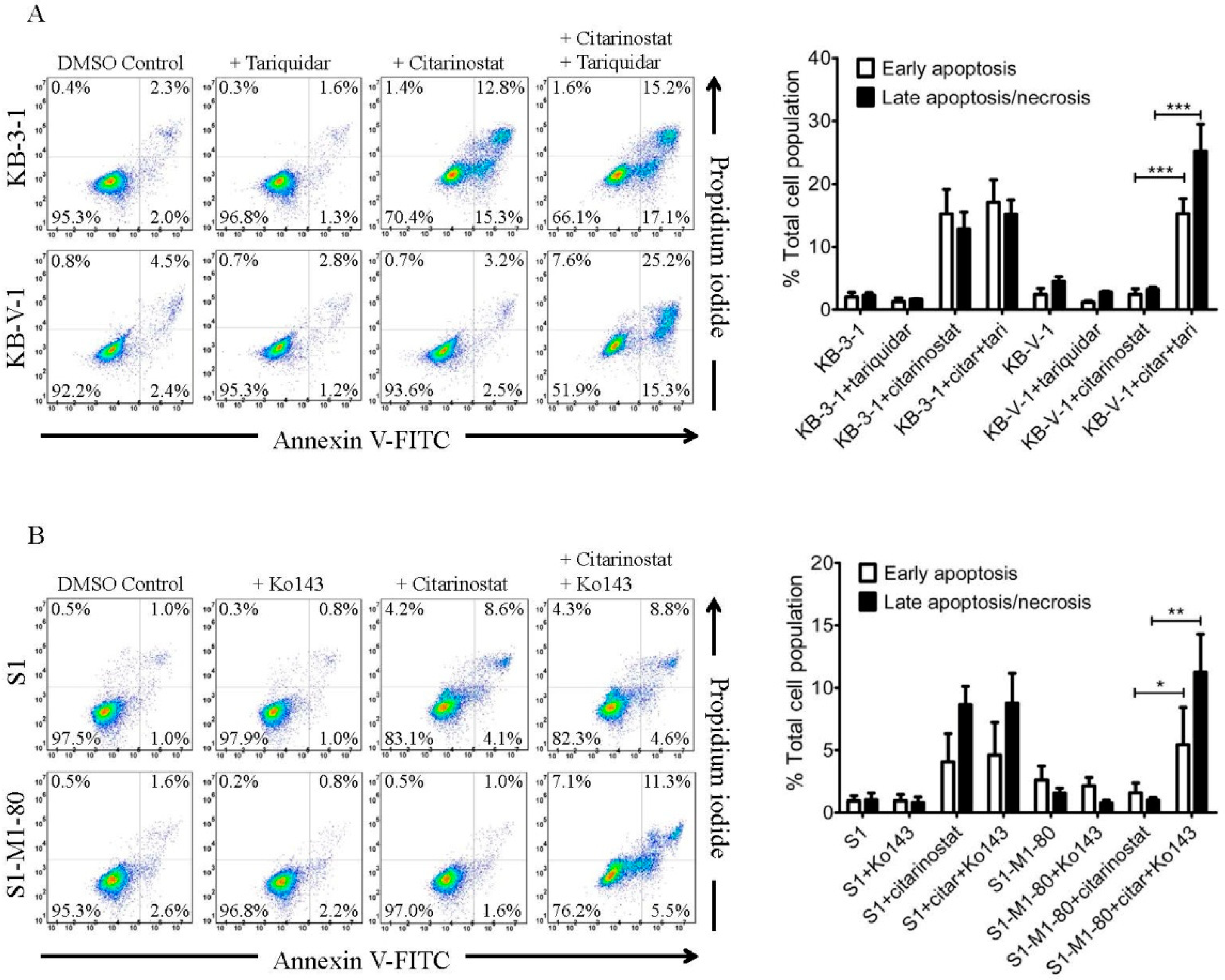
4.5. Apoptosis Assays
4.6. Citarinostat Accumulation Assay and HPLC-MS/MS Analysis
4.7. Docking of Citarinostat in the Substrate-Binding Pocket of Human ABCB1 and ABCG2
4.8. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [Green Version]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2009, 4, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, M.; Younes, A. Histone deacetylase inhibitors in the treatment of lymphoma. Discov. Med. 2010, 10, 462–470. [Google Scholar]
- Fraczek, J.; Vanhaecke, T.; Rogiers, V. Toxicological and metabolic considerations for histone deacetylase inhibitors. Expert Opin. Drug Metab. Toxicol. 2013, 9, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Moreau, P.; Laubach, J.P.; Maglio, M.E.; Lonial, S.; San-Miguel, J. Deacetylase inhibitors as a novel modality in the treatment of multiple myeloma. Pharm. Res. 2017, 117, 185–191. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; George, T.I.; Linder, A.; Langford, C.; Perkins, C.; Ma, J.; Westervelt, P.; Merker, J.D.; Berube, C.; Coutre, S.; et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia 2018, 32, 470–478. [Google Scholar] [CrossRef]
- Cengiz Seval, G.; Beksac, M. A comparative safety review of histone deacetylase inhibitors for the treatment of myeloma. Expert Opin. Drug Saf. 2019, 18, 563–571. [Google Scholar] [CrossRef]
- Borrelli, E.P.; McGladrigan, C.G. Differences in safety profiles of newly approved medications for multiple myeloma in real-world settings versus randomized controlled trials. J. Oncol. Pharm. Pract. 2020, 1–10. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Tapadar, S.; Luchini, D.N.; Kim, K.H.; Billadeau, D.D. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: A new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J. Med. Chem. 2008, 51, 4370–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalin, J.H.; Bergman, J.A. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem. 2013, 56, 6297–6313. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Wagner, F.F.; Kaya, T.; Gale, J.P.; Aidoud, N.; Davoine, E.L.; Lazzaro, F.; Weiwer, M.; Zhang, Y.L.; Holson, E.B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013, 56, 4816–4820. [Google Scholar] [CrossRef]
- Thaler, F.; Mercurio, C. Towards selective inhibition of histone deacetylase isoforms: What has been achieved, where we are and what will be next. ChemMedChem 2014, 9, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wong, J.C.; Zhang, W.; Wang, Z.; Zhang, N.; Peng, Z.; Zhang, Z.; Rong, Y.; Li, S.; Zhang, M.; et al. Identification of a novel aminotetralin class of HDAC6 and HDAC8 selective inhibitors. J. Med. Chem. 2014, 57, 8026–8034. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chen, W.; Qiu, Z.; Guo, L.; Zhu, W.; Li, W.; Wang, Z.; Zhang, W.; Zhang, Z.; Rong, Y.; et al. Design and synthesis of orally bioavailable aminopyrrolidinone histone deacetylase 6 inhibitors. J. Med. Chem. 2015, 58, 2809–2820. [Google Scholar] [CrossRef]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, e0173507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kekatpure, V.D.; Dannenberg, A.J.; Subbaramaiah, K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem. 2009, 284, 7436–7445. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Das, D.S.; Song, Y.; Hideshima, T.; Tai, Y.T.; Chauhan, D.; Anderson, K.C. Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2018, 32, 843–846. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Du, T.; Chauhan, D.; Anderson, K.C. Preclinical validation of Alpha-Enolase (ENO1) as a novel immunometabolic target in multiple myeloma. Oncogene 2020, 39, 2786–2796. [Google Scholar] [CrossRef]
- Cosenza, M.; Civallero, M.; Marcheselli, L.; Sacchi, S.; Pozzi, S. Citarinostat and Momelotinib co-target HDAC6 and JAK2/STAT3 in lymphoid malignant cell lines: A potential new therapeutic combination. Apoptosis Int. J. Program. Cell Death 2020, 25, 370–387. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guerrero, E.; Gotz, R.; Doose, S.; Sauer, M.; Rodriguez-Gil, A.; Nerreter, T.; Kortum, K.M.; Perez-Simon, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2020, 35, 201–214. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, S.W.; Jeon, Y.H.; Lee, D.H.; Kim, G.W.; Yoo, J.; Kim, S.Y.; Kwon, S.H. Combination of ACY-241 and JQ1 Synergistically Suppresses Metastasis of HNSCC via Regulation of MMP-2 and MMP-9. Int. J. Mol. Sci. 2020, 21, 6873. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Brozik, A.; Hegedus, C.; Erdei, Z.; Hegedus, T.; Ozvegy-Laczka, C.; Szakacs, G.; Sarkadi, B. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: Substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin. Drug Metab. Toxicol. 2011, 7, 623–642. [Google Scholar] [CrossRef]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Noguchi, K.; Katayama, K.; Sugimoto, Y. Human ABC transporter ABCG2/BCRP expression in chemoresistance: Basic and clinical perspectives for molecular cancer therapeutics. Pharm. Pers. Med. 2014, 7, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Hsieh, C.H.; Wu, Y.S. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol. Pharm. 2011, 8, 1996–2011. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, L.M.; Belch, A.R. Intrinsic expression of the multidrug transporter, P-glycoprotein 170, in multiple myeloma: Implications for treatment. Leuk Lymphoma 1995, 17, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, L.M.; Szczepek, A.J.; Belch, A.R. Deficient drug transporter function of bone marrow-localized and leukemic plasma cells in multiple myeloma. Blood 1997, 90, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H. Expression of MDR1/P-glycoprotein, the multidrug resistance protein MRP, and the lung-resistance protein LRP in multiple myeloma. Med. Oncol. 2002, 19, 87–104. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Abe, S.; Kurata, M.; Hasegawa, M.; Yamamoto, K.; Inoue, M.; Takemura, T.; Suzuki, K.; Kitagawa, M. IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am. J. Hematol. 2006, 81, 824–831. [Google Scholar] [CrossRef]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Komai, M.; Nishinobo, M.; Yamashita, M.; Yanae, M.; Yamazoe, Y.; Nishida, S. Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk Res. 2012, 36, 1315–1322. [Google Scholar] [CrossRef]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef]
- Matthews, C.; Catherwood, M.A.; Larkin, A.M.; Clynes, M.; Morris, T.C.; Alexander, H.D. MDR-1, but not MDR-3 gene expression, is associated with unmutated IgVH genes and poor prognosis chromosomal aberrations in chronic lymphocytic leukemia. Leuk Lymphoma 2006, 47, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.D.; Karp, J.E.; Chen, T.T.; Doyle, L.A. Expression of breast cancer resistance protein in blast cells from patients with acute leukemia. Blood 2000, 96, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, D.; Sell, W.; Voigt, A.; Hermann, J.; Zintl, F.; Sauerbrey, A. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia 2002, 16, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Uggla, B.; Stahl, E.; Wagsater, D.; Paul, C.; Karlsson, M.G.; Sirsjo, A.; Tidefelt, U. BCRP mRNA expression v. clinical outcome in 40 adult AML patients. Leuk Res. 2005, 29, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, A.A.; Tsvetaeva, D.A.; Grudinskaja, T.V. Role of ABC-cassette transporters (MDR1, MRP1, BCRP) in the development of primary and acquired multiple drug resistance in patients with early and metastatic breast cancer. Exp. Oncol. 2013, 35, 287–290. [Google Scholar] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, T.; Shiozawa, K.; Hassel, B.A.; Ross, D.D. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood 2006, 108, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, C.; Ozvegy-Laczka, C.; Szakacs, G.; Sarkadi, B. Interaction of ABC multidrug transporters with anticancer protein kinase inhibitors: Substrates and/or inhibitors? Curr. Cancer Drug Targets 2009, 9, 252–272. [Google Scholar]
- Wu, C.P.; Sim, H.M.; Huang, Y.H.; Liu, Y.C.; Hsiao, S.H.; Cheng, H.W.; Li, Y.Q.; Ambudkar, S.V.; Hsu, S.C. Overexpression of ATP-binding cassette transporter ABCG2 as a potential mechanism of acquired resistance to vemurafenib in BRAF(V600E) mutant cancer cells. Biochem. Pharm. 2013, 85, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.P.; Hsiao, S.H.; Su, C.Y.; Luo, S.Y.; Li, Y.Q.; Huang, Y.H.; Hsieh, C.H.; Huang, C.W. Human ATP-Binding Cassette transporters ABCB1 and ABCG2 confer resistance to CUDC-101, a multi-acting inhibitor of histone deacetylase, epidermal growth factor receptor and human epidermal growth factor receptor 2. Biochem. Pharm. 2014, 92, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Hsieh, C.H.; Hsiao, S.H.; Luo, S.Y.; Su, C.Y.; Li, Y.Q.; Huang, Y.H.; Huang, C.W.; Hsu, S.C. Human ATP-Binding Cassette Transporter ABCB1 Confers Resistance to Volasertib (BI 6727), a Selective Inhibitor of Polo-like Kinase 1. Mol. Pharm. 2015, 12, 3885–3895. [Google Scholar] [CrossRef]
- Estiu, G.; Greenberg, E.; Harrison, C.B.; Kwiatkowski, N.P.; Mazitschek, R.; Bradner, J.E.; Wiest, O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51, 2898–2906. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Hsieh, Y.J.; Murakami, M.; Vahedi, S.; Hsiao, S.H.; Yeh, N.; Chou, A.W.; Li, Y.Q.; Wu, Y.S.; Yu, J.S.; et al. Human ATP-binding cassette transporters ABCB1 and ABCG2 confer resistance to histone deacetylase 6 inhibitor ricolinostat (ACY-1215) in cancer cell lines. Biochem. Pharm. 2018, 155, 316–325. [Google Scholar] [CrossRef]
- Wu, C.P.; Hung, C.Y.; Lusvarghi, S.; Huang, Y.H.; Tseng, P.J.; Hung, T.H.; Yu, J.S.; Ambudkar, S.V. Overexpression of ABCB1 and ABCG2 contributes to reduced efficacy of the PI3K/mTOR inhibitor samotolisib (LY3023414) in cancer cell lines. Biochem. Pharm. 2020, 180, 114137. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Cheng, M.; Cai, W.; Huang, W.; Chen, Y.; Wu, Z.; Luo, P.; Yan, W. Histone deacetylase 6 regulated expression of IL-8 is involved in the doxorubicin (Dox) resistance of osteosarcoma cells via modulating ABCB1 transcription. Eur. J. Pharm. 2018, 840, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased mdr1 expression can precede gene amplification. Science 1986, 232, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, D.A.; Monks, A.; Sausville, E.A. Cell line designation change: Multidrug-resistant cell line in the NCI anticancer screen. J. Natl. Cancer Inst. 1998, 90, 862. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.P.; Shukla, S.; Calcagno, A.M.; Hall, M.D.; Gottesman, M.M.; Ambudkar, S.V. Evidence for dual mode of action of a thiosemicarbazone, NSC73306: A potent substrate of the multidrug resistance linked ABCG2 transporter. Mol. Cancer 2007, 6, 3287–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar] [PubMed]
- Henrich, C.J.; Bokesch, H.R.; Dean, M.; Bates, S.E.; Robey, R.W.; Goncharova, E.I.; Wilson, J.A.; McMahon, J.B. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J. Biomol. Screen. 2006, 11, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, M.; Tominaga, H.; Shiga, M.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol. Pharm. Bull. 1996, 19, 1518–1520. [Google Scholar] [CrossRef] [Green Version]
- Anderson, H.A.; Maylock, C.A.; Williams, J.A.; Paweletz, C.P.; Shu, H.; Shacter, E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 2003, 4, 87–91. [Google Scholar] [CrossRef]
- Wu, C.P.; Hsieh, Y.J.; Hsiao, S.H.; Su, C.Y.; Li, Y.Q.; Huang, Y.H.; Huang, C.W.; Hsieh, C.H.; Yu, J.S.; Wu, Y.S. Human ATP-Binding Cassette Transporter ABCG2 Confers Resistance to CUDC-907, a Dual Inhibitor of Histone Deacetylase and Phosphatidylinositol 3-Kinase. Mol. Pharm. 2016, 13, 784–794. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abhold, E.L.; Kiang, A.; Rahimy, E.; Kuo, S.Z.; Wang-Rodriguez, J.; Lopez, J.P.; Blair, K.J.; Yu, M.A.; Haas, M.; Brumund, K.T.; et al. EGFR kinase promotes acquisition of stem cell-like properties: A potential therapeutic target in head and neck squamous cell carcinoma stem cells. PLoS ONE 2012, 7, e32459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Mean IC50 ± SD [μM] 1 (R.F 2) | ||
|---|---|---|---|
| Citarinostat | Citarinostat + Tariquidar | Citarinostat + Ko143 | |
| KB-3-1 | 3.98 ± 0.73 (1) | 4.59 ± 0.67 (1) | N.D |
| KB-V-1 | 57.96 ± 5.56 (15) | 3.01 ± 0.57 *** (1) | N.D |
| OVCAR-8 | 7.52 ± 1.10 (1) | 7.46 ± 1.55 (1) | N.D |
| NCI-ADR-RES | 84.15 ± 9.91 (11) | 5.56 ± 1.10 *** (1) | N.D |
| S1 | 2.82 ± 0.40 (1) | N.D | 2.75 ± 0.45 (1) |
| S1-M1-80 | 59.66 ± 10.32 (21) | N.D | 2.94 ± 0.50 *** (1) |
| H460 | 7.46 ± 0.36 (1) | N.D | 6.17 ± 0.53 * (1) |
| H460-MX20 | 23.70 ± 1.58 (3) | N.D | 6.08 ± 0.36 *** (1) |
| pcDNA-HEK293 | 3.01 ± 0.44 (1) | 2.18 ± 0.21 (1) | 1.57 ± 0.20 ** (1) |
| MDR19-HEK293 | 33.83 ± 3.21 (11) | 3.19 ± 0.21 *** (1) | N.D |
| R482-HEK293 | 20.11 ± 2.48 *** (7) | N.D | 4.78 ± 0.37 *** (3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-P.; Hung, C.-Y.; Lusvarghi, S.; Chang, Y.-F.; Hsiao, S.-H.; Huang, Y.-H.; Hung, T.-H.; Yu, J.-S.; Ambudkar, S.V. Overexpression of Human ABCB1 and ABCG2 Reduces the Susceptibility of Cancer Cells to the Histone Deacetylase 6-Specific Inhibitor Citarinostat. Int. J. Mol. Sci. 2021, 22, 2592. https://doi.org/10.3390/ijms22052592
Wu C-P, Hung C-Y, Lusvarghi S, Chang Y-F, Hsiao S-H, Huang Y-H, Hung T-H, Yu J-S, Ambudkar SV. Overexpression of Human ABCB1 and ABCG2 Reduces the Susceptibility of Cancer Cells to the Histone Deacetylase 6-Specific Inhibitor Citarinostat. International Journal of Molecular Sciences. 2021; 22(5):2592. https://doi.org/10.3390/ijms22052592
Chicago/Turabian StyleWu, Chung-Pu, Cheng-Yu Hung, Sabrina Lusvarghi, Yen-Fu Chang, Sung-Han Hsiao, Yang-Hui Huang, Tai-Ho Hung, Jau-Song Yu, and Suresh. V. Ambudkar. 2021. "Overexpression of Human ABCB1 and ABCG2 Reduces the Susceptibility of Cancer Cells to the Histone Deacetylase 6-Specific Inhibitor Citarinostat" International Journal of Molecular Sciences 22, no. 5: 2592. https://doi.org/10.3390/ijms22052592