
Multifaceted Mechanisms of Action of Metformin Which Have Been Unraveled One after Another in the Long History


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glucose Toxicity Is an Underlying Mechanism for Type 2 Diabetes Mellitus
3. Various Agents for Type 2 Diabetes Mellitus Protect Pancreatic β-Cells against Glucose Toxicity
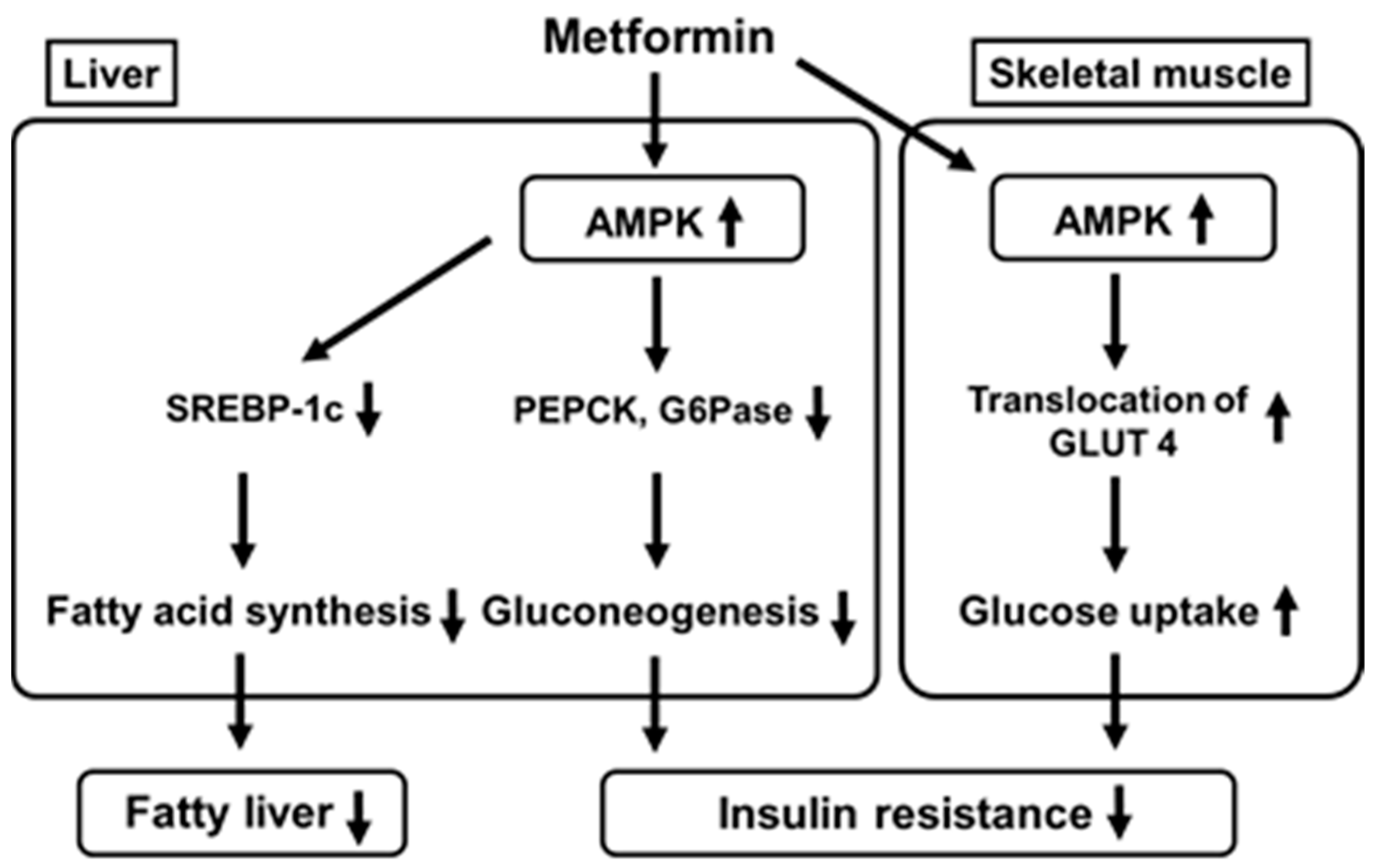
4. Metformin Activates Adenosine Monophosphate-Activated Protein Kinase (AMPK) in the Liver and Skeletal Muscle Which Leads to Suppression of Gluconeogenesis in the Liver and Increase in Glucose Uptake into Skeletal Muscle
5. Metformin Suppresses Glucagon Signaling in the Liver by Suppressing Adenylate Cyclase Which Leads to Suppression of Gluconeogenesis in the Liver
6. Metformin Reduces Autophagy Failure Observed in Pancreatic β-Cells under Diabetic Conditions
7. Metformin Alters the Gut Microbiome and Glucose Absorption from the Intestine
8. Metformin Reduces Food Intake and Lowers Body Weight by Increasing Circulating Level of the Peptide Hormone Growth/Differentiation Factor 15 (GDF15)
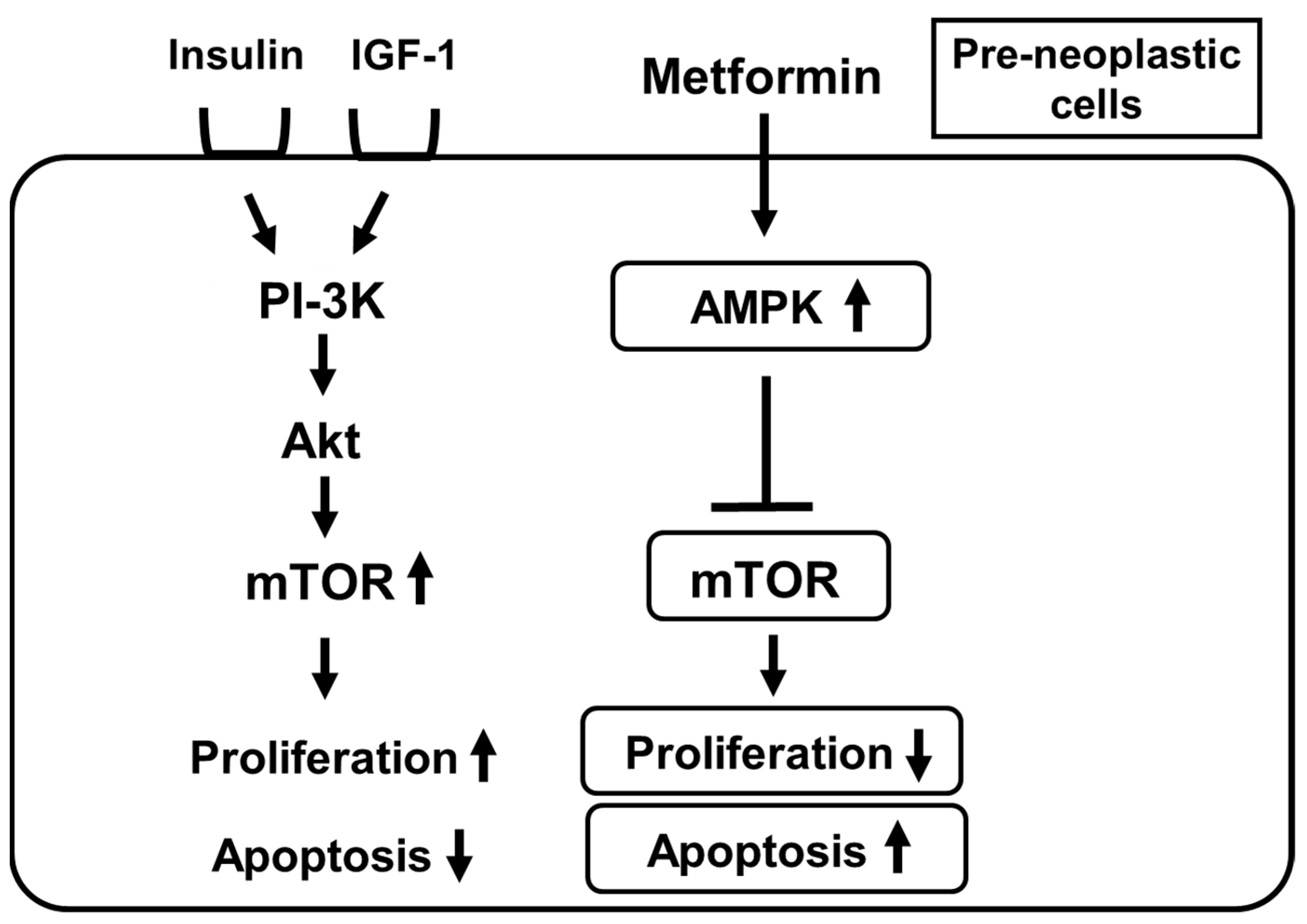
9. Metformin Suppresses Mechanistic Target of Rapamycin (Mtor) by Activating AMPK in Pre-Neoplastic Cells and Thereby Suppresses the Onset and/or Development of Various Cancers
10. Metformin Consumption Potentially Influences the Mortality in Subjects with Type 2 Diabetes Mellitus and Coronavirus Infectious Disease (COVID-19)
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. β-Cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50, S154–S159. [Google Scholar] [CrossRef] [Green Version]
- Poitout, V.; Robertson, R.P. Minireview: Secondary beta cell failure in type 2 diabetes: A convergence of glucotoxicity and lipotoxicity. Endocrinology 2001, 143, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J. Type 2 diabetes-a matter of beta-cell life and death? Science 2005, 307, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Matsuoka, T.; Kimura, T.; Obata, A.; Shimoda, M.; Kamei, S.; Mune, T.; Kaku, K. Appropriate therapy for type 2 diabetes in view of pancreatic β-cell glucose toxicity: “The earlier, the better”. J. Diabetes 2016, 8, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Obata, A.; Kimura, T.; Shimoda, M.; Sanada, J.; Fushimi, Y.; Katakami, N.; Matsuoka, T.; Kaku, K. Notable underlying mechanism for pancreatic β-cell dysfunction and atherosclerosis: Pleiotropic role of incretin and insulin signaling in various situation. Int. J. Mol. Sci. 2020, 21, 9444. [Google Scholar] [CrossRef]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic β-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Matsuoka, T.; Kawashima, S.; Takebe, S.; Kubo, N.; Miyatsuka, T.; Kaneto, H.; Shimomura, I. A novel function of Onecut 1 as a negative regulator of MafA. J. Biol. Chem. 2013, 288, 21648–21658. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving MafA expression in diabetic islet β-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Matsuoka, T. Role of pancreatic transcription factors in maintenance of mature -cell function. Int. J. Mol. Sci. 2015, 16, 6281–6297. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, W.; Takahashi, S.; Yasuda, K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 2015, 58, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. β-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.M.; Hale, M.A.; Kagami, H.; Hammer, R.E.; MacDonald, R.J. Experimental control of pancreatic development and maintenance. Proc. Natl. Acad. Sci. USA 2002, 99, 12236–12241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018. [Google Scholar] [CrossRef] [Green Version]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 2006, 281, 1091–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Miyatsuka, T.; Sasaki, S.; Miyashita, K.; Kubo, N.; Shimo, N.; Takebe, S.; Watada, H.; Kaneto, H.; Matsuoka, T.; et al. Recovered expression of Pdx1 improves β-cell failure in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 483, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Kaneto, H.; Laybutt, D.R.; Duvivier-Kali, V.; Trivedi, N.; Suzuma, K.; King, G.L.; Weir, G.C.; Bonner-Weir, S. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: Possible contribution to the impaired incretin effects in diabetes. Diabetes 2007, 56, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, F.; Miyatsuka, T.; Sasaki, S.; Takahara, M.; Yamamoto, Y.; Shimo, N.; Watada, H.; Kaneto, H.; Gannon, M.; Matsuoka, T.; et al. Sustained expression of GLP-1 receptor differentially modulates β-cell functions in diabetic and nondiabetic mice. Biochem. Biophys. Res. Commun. 2016, 471, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.H.; McIntosh, C.H.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399. [Google Scholar] [CrossRef]
- Liu, Z.; Habener, J.F. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 2008, 283, 8723–8735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamoto, I.; Kubota, N.; Nakaya, K.; Kumagai, K.; Hashimoto, S.; Kubota, T.; Inoue, M.; Kajiwara, E.; Katsuyama, H.; Obata, A.; et al. TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia 2014, 57, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, R.K.; Mondragon, A.; Chen, L.; McGinty, J.A.; French, P.M.; Ferrer, J.; Thorens, B.; Hodson, D.J.; Rutter, G.A.; Xavier, G.D. Selective disruption of Tcf7l2 in the pancreatic β cell impairs secretory function and lowers β cell mass. Hum. Mol. Genet. 2015, 24, 1390–1399. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Vicent, D.; Suzuma, K. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J. Clin. Investig. 2003, 111, 1835–1842. [Google Scholar] [CrossRef] [Green Version]
- Mukai, Y.; Rikitake, Y.; Shiojima, I.; Wolfrum, S.; Satoh, M.; Takeshita, K.; Hiroi, Y.; Salomone, S.; Kim, H.H.; Benjamin, L.E.; et al. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J. Clin. Investig. 2006, 116, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Kubota, N.; Sato, H.; Sasaki, M.; Takamoto, I.; Kubota, T.; Nakaya, K.; Noda, M.; Ueki, K.; Kadowaki, T. Insulin receptor substrate-2 (Irs2) in endothelial cells plays a crucial role in insulin secretion. Diabetes 2015, 64, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Obata, A.; Kimura, T.; Obata, Y.; Shimoda, M.; Kinoshita, T.; Kohara, K.; Okauchi, S.; Hirukawa, H.; Kamei, S.; Nakanishi, S.; et al. Vascular endothelial PDK1 plays pivotal roles for maintenance of pancreatic beta-cell mass and function in adult male mice. Diabetologia 2019, 62, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, S.; Matsuoka, T.; Kaneto, H.; Tochino, Y.; Kato, K.; Yamamoto, K.; Yamamoto, T.; Matsuhisa, M.; Shimomura, I. Effect of alogliptin, pioglitazone and glargine on pancreatic β-cells in diabetic db/db mice. Biochem. Biophys. Res. Commun. 2011, 404, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Kanda, Y.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Matsuki, M.; Kaku, K. The human glucagon-like peptide-1 analogue liraglutide preserves pancreatic beta cells via regulation of cell kinetics and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetologia 2011, 54, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, S.; Kanda, Y.; Shimoda, M.; Tatsumi, F.; Kohara, K.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Vildagliptin preserves the mass and function of pancreatic beta cells via the developmental regulation and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetes Obes. Metab. 2013, 15, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Hirukawa, H.; Kaneto, H.; Shimoda, M.; Kimura, T.; Okauchi, S.; Obata, A.; Kohara, K.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; et al. Combination of DPP-4 inhibitor and PPARγ agonist exerts protective effects on pancreatic β-cells in diabetic db/db mice through the augmentation of IRS-2 expression. Mol. Cell. Endocrinol. 2015, 413, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kaneto, H.; Shimoda, M.; Hirukawa, H.; Hamamoto, S.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Protective effects of pioglitazone and/or liraglutide on pancreatic β-cells: Comparison of their effects between in an early and advanced stage of diabetes. Mol. Cell. Endocrinol. 2015, 400, 78–89. [Google Scholar] [CrossRef]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Goto, H.; Nomiyama, T.; Mita, T.; Yasunari, E.; Azuma, K.; Komiya, K.; Arakawa, M.; Jin, W.L.; Kanazawa, A.; Kawamori, R.; et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces intimal thickening after vascular injury. Biochem. Biophys. Res. Commun. 2011, 405, 79–84. [Google Scholar] [CrossRef]
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor Mediates Cardiovascular Protection by Liraglutide In Mice With Experimental Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Obata, A.; Shimoda, M.; Okauchi, S.; Hirukawa, H.; Kohara, K.; Kinoshita, T.; Nogami, Y.; Nakanishi, S.; Mune, T.; et al. Decreased GLP-1 receptor expression in endothelial and smooth muscle cells in diabetic db/db mice: TCF7L2 is a possible regulator of vascular GLP-1 receptor. Diabetes Vasc. Dis. Res. 2017, 14, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Obata, A.; Shimoda, M.; Shimizu, I.; da Silva Xavier, G.; Okauchi, S.; Hirukawa, H.; Kohara, K.; Mune, T.; Moriuchi, S.; et al. Down-regulation of vascular GLP-1 receptor expression in human subjects with obesity. Sci. Rep. 2018, 8, 10644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, J.; Obata, A.; Obata, Y.; Fushiimi, Y.; Shimoda, M.; Kohara, K.; Nakanishi, S.; Mune, T.; Kaku, K.; Kaneto, H. Dulaglutide exerts beneficial anti-atherosclerotic effects in ApoE knockout mice with diabetes: The earlier, the better. Sci. Rep. 2021, 11, 1425. [Google Scholar] [CrossRef] [PubMed]
- Mita, T.; Katakami, N.; Yoshii, H.; Onuma, T.; Kaneto, H.; Osonoi, T.; Shiraiwa, T.; Kosugi, K.; Umayahara, Y.; Yamamoto, T.; et al. Alogliptin, a dipeptidyl peptidase-4 inhibitor, prevents the progression of carotid Atherosclerosis in patients with type 2 diabetes mellitus: The Study of Preventive Effects of Alogliptin on Diabetic Atherosclerosis (SPEAD-A). Diabetes Care 2016, 39, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Mita, T.; Katakami, N.; Shiraiwa, T.; Yoshii, H.; Onuma, T.; Kuribayashi, N.; Osonoi, T.; Kaneto, H.; Kosugi, K.; Umayahara, Y.; et al. Sitagliptin attenuates the progression of carotid intima-media thickening in insulin-treated patients with type 2 diabetes mellitus: The Sitagliptin Preventive study of Intima-media thickness Evaluation (SPIKE). Diabetes Care 2016, 39, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Poulter, N.R.; Bhatt, D.L.; Bain, S.C.; Buse, J.B.; Leiter, L.A.; Nauck, M.A.; Pratley, R.E.; Zinman, B.; Ørsted, D.D.; et al. Effects of liraglutide on cardiovascular outcomes in patients with type 2 diabetes mellitus with or without history of myocardial infarction or stroke. Circulation 2018, 138, 2884–2894. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, S. Mitigating cardiovascular risk in type 2 diabetes with antidiabetes drugs: A review of principal cardiovascular outcome results of EMPA-REG OUTCOME, LEADER, and SUSTAIN-6 Trials. Diabetes Care 2017, 40, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Ryden, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2017, 394, 121–130. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rorth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Kober, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Husain, M.; Bain, S.C.; Jeppesen, O.K.; Lingvay, I.; Sorrig, R.; Treppendahl, M.B.; Vilsbøll, T. Semaglutide (SUSTAIN and PIONEER) reduces cardiovascular events in type 2 diabetes across varying cardiovascular risk. Diabetes Obes. Metab. 2020, 22, 442–451. [Google Scholar] [CrossRef]
- Iqbal, A.M.; Imamudeen, N.; Basheer, A.; Menon, S.; Mohan, G.; Sani, T.N.; Haroon, N.N. Efficacy and Cardiovascular Safety of GLP-1 Receptor Analogues. Curr. Drug Saf. 2020. [Google Scholar] [CrossRef]
- Smith, S.A. Central role of the adipocyte in the insulin-sensitising and cardiovascular risk modifying actions of the thiazolidinediones. Biochimie 2003, 85, 1219–1230. [Google Scholar] [CrossRef]
- Bailey, C.J. Treating insulin resistance in type 2 diabetes with metformin and thiazolidinediones. Diabetes Obes. Metab. 2005, 7, 675–691. [Google Scholar] [CrossRef]
- Shimo, N.; Matsuoka, T.; Miyatsuka, T.; Takebe, S.; Tochino, Y.; Takahara, M.; Kaneto, H.; Shimomura, I. Short-term selective alleviation of glucotoxicity and lipotoxicity ameliorates the suppressed expression of key β-cell factors under diabetic conditions. Biochem. Biophys. Res. Commun. 2015, 467, 948–954. [Google Scholar] [CrossRef]
- Okauchi, S.; Shimoda, M.; Obata, A.; Kimura, T.; Hirukawa, H.; Kohara, K.; Mune, T.; Kaku, K.; Kaneto, H. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic β-cells in obese type 2 diabetic db/db mice. Biochem. Biophys. Res. Commun. 2016, 470, 772–782. [Google Scholar] [CrossRef]
- Kimura, T.; Obata, A.; Shimoda, M.; Okauchi, S.; Kanda-Kimura, Y.; Nogami, Y.; Hirukawa, H.; Kohara, K.; Nakanishi, S.; Mune, T.; et al. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic β-cells in obese diabetic db/db mice: The earlier and longer, the better. Diabetes Obes. Metab. 2018, 20, 2442–2457. [Google Scholar] [CrossRef]
- Cefalu, W.T. Paradoxical insights into whole body metabolic adaptations following SGLT2 inhibition. J. Clin. Investig. 2014, 124, 485–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Fiorentino, T.V.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514. [Google Scholar] [CrossRef]
- Obata, A.; Kubota, N.; Kubota, T.; Iwamoto, M.; Sato, H.; Sakurai, Y.; Takamoto, I.; Katsuyama, H.; Suzuki, Y.; Fukazawa, M.; et al. Tofogliflozin improves insulin resistance in skeletal muscle and accelerates lipolysis in adipose tissue in male mice. Endocrinology 2016, 157, 1029–1042. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamii, T.; Nogami, M.; Ogawa, W. Mechanisms of metformin action: In and out of the gut. J. Diabetes Investig. 2018, 9, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Fujitani, Y.; Ebato, C.; Uchida, T.; Kawamori, R.; Watada, H. β-cell autophagy: A novel mechanism regulating beta-cell function and mass: Lessons from b-cell-specific Atg7-deficient mice. Islets 2009, 1, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masini, M.; Lupi, R.; Bugliani, M.; Boggi, U.; Filipponi, F.; Masiello, P.; Marchetti, P. A role for autophagy in β-cell life and death. Islets 2009, 1, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Bartolome, A.; Kimuta-Koyanagi, M.; Asahara, S.-I.; Guillen, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; Garcia-Aguilar, A.; Koike, M.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef] [Green Version]
- Watada, H.; Fujitani, Y. Minireview: Autophagy in pancreatic β-cells and its implication in diabetes. Mol. Endocrinol. 2015, 29, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Nogami, M.; Sakaguchi, K.; Okada, Y.; Hirota, Y.; Sugawara, K.; Tamori, Y.; Zeng, F.; Murakami, T.; Ogawa, W. Enhanced Release of Glucose into the Intraluminal Space of the Intestine Associated With Metformin Treatment as Revealed by [18F] Fluorodeoxyglucose PET-MRI. Diabetes Care 2020, 43, 1796–1802. [Google Scholar] [CrossRef]
- Ito, J.; Nogami, M.; Morita, Y.; Sakaguchi, K.; Komada, H.; Hirota, Y.; Sugawara, K.; Tamori, Y.; Zeng, F.; Murakami, T.; et al. Dose-dependent accumulation of glucose in the intestinal wall and lumen induced by metformin as revealed by [18F]-labelled fluorodeoxyglucose positron emission tomography-MRI. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef]
- Yang, L.; Chang, C.C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjær, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Crawley, S.; Chen, M.; Ayupova, D.A.; Lindhout, D.A.; Higbee, J.; Kutach, A.; Joo, W.; Gao, Z.; Fu, D.; et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 2017, 550, 255–259. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Pare, G.; Hess, S.; Ford, R.J.; Sjaarda, J.; Raman, K.; McQueen, M.; Lee, S.; Haenel, H.; Steinberg, G.R.; et al. Growth differentiation factor 15 as a novel biomarker for metformin. Diabetes Care 2017, 40, 280–283. [Google Scholar] [CrossRef] [Green Version]
- Cimino, I.; Coll, A.P.; Yeo, G.S.H. GDF15 and energy balance: Homing in on a mechanism. Nat. Med. 2017, 23, 1119–1120. [Google Scholar] [CrossRef]
- Tsai, V.W.W.; Husaini, Y.; Sainsbury, A.; Brown, D.A.; Breit, S.N. The MIC-1/GDF15-GFRAL pathway in energy homeostasis: Implications for obesity, cachexia, and other associated diseases. Cell Metab. 2018, 28, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Apolzan, J.W.; Venditti, E.M.; Edelstein, S.L.; Knowler, W.C.; Dabelea, D.; Boyko, E.J.; Pi-Sunyer, X.; Kalyani, R.R.; Franks, P.W.; Srikanthan, P.; et al. Long-term weight loss with metformin or lifestyle intervention in the diabetes prevention program outcomes study. Ann. Intern. Med. 2019, 170, 682–690. [Google Scholar] [CrossRef]
- Patel, S.; Alvarez-Guaita, A.; Melvin, A.; Rimmington, D.; Dattilo, A.; Miedzybrodzka, E.L.; Cimino, I.; Maurin, A.C.; Roberts, G.P.; Meek, C.L.; et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 2019, 29, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 2020, 578, 444–448. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer. 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Mayor, S. High glucose and diabetes increase cancer risk. Lancet Oncol. 2005, 6, 71. [Google Scholar] [CrossRef]
- Noto, H.; Goto, S.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef] [PubMed]
- Noto, H.; Goto, A.; Tsujimoto, T.; Osame, K.; Noda, M. Latest insights into the risk of cancer in diabetes. J. Diabetes. Investig. 2013, 4, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.J.; Johnson, J.A.; Wild, S.H. Diabetes treatments and cancer risk: The importance of considering aspects of drug exposure. Lancet Diabetes Endocrinol. 2013, 1, 132–139. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, F.B. The global implications of diabetes and cancer. Lancet 2014, 383, 1947–1948. [Google Scholar] [CrossRef]
- Rahman, A. Type 2 diabetes and risk of pancreatic adenocarcinoma. Lancet Oncol. 2014, 15, e420. [Google Scholar] [CrossRef]
- Gregg, E.W.; Cheng, Y.J.; Srinivasan, M.; Lin, J.; Geiss, L.S.; Albright, A.L.; Imperatore, G. Trends in cause-specific mortality among adults with and without diagnosed diabetes in the USA: An epidemiological analysis of linked national survey and vital statistics data. Lancet 2018, 391, 2430–2440. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [Green Version]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New application for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Chan, A.T. Metformin for cancer prevention: A reason for optimism. Lancet Oncol. 2016, 17, 407–409. [Google Scholar] [CrossRef]
- Demb, J.; Yaseyyedi, A.; Liu, L.; Bustamante, R.; Earles, A.; Ghosh, P.; Gutkind, J.S.; Gawron, A.J.; Kaltenbach, T.R.; Martinez, M.E.; et al. Metformin is associated with reduced odds for colorectal cancer among persons with dDiabetes. Clin. Trans. Gastroenterol. 2019, 10, e00092. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Q.; Zhou, X.C.; Du, P.; Yin, M.Y.; Xu, L.; Chen, W.J.; Xu, C.F. Relationships are between metformin use and survival in pancreatic cancer patients concurrent with diabetes: A systematic review and meta-analysis. Medicine 2020, 99, e21687. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, E.A.; Lee, J.W.; Kim, Y.; You, H.S.; Han, Y.E.; Kim, H.S.; Bae, Y.J.; Kang, H.T.; Kim, J. Metformin use reduced the overall risk of cancer in diabetic patients: A study based on the Korean NHIS-HEALS cohort. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1714–1722. [Google Scholar] [CrossRef]
- Lee, J.-W.; Choi, E.-A.; Kim, Y.-S.; Kim, Y.; You, H.-S.; Han, Y.-E.; Kim, H.-P.; Bae, Y.-J.; Kim, J.; Kang, H.-T. Metformin usage and the risk of colorectal cancer: A national cohort study. Int. J. Colorectal Dis. 2021, 36, 303–310. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Huang, I.; Lim, M.A.; Pranata, R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumoniae: A systematic review, meta-analysis, and meta-regression. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Lukito, A.A.; Pranata, R.; Henrina, J.; Lim, M.A.; Lawrensia, S.; Suastika, K. The Effect of Metformin Consumption on Mortality in Hospitalized COVID-19 patients: A systematic review and meta-analysis. Diabetes Metab. Syndr. 2020, 14, 2177–2183. [Google Scholar] [CrossRef]
- Penlioglou, T.; Papachristou, S.; Papanas, N. COVID-19 and Diabetes Mellitus: May Old Anti-diabetic Agents Become the New Philosopher’s Stone? Diabetes Ther. 2020, 11, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hadjadj, S.; Wargny, M.; Pichelin, M.; Al-Salameh, A.; Allix, I.; Amadou, C.; Arnault, G.; Baudoux, F.; Bauduceau, B.; et al. Phenotypic characteristics and prognosis of inpatients with COVID-19 and diabetes: The CORONADO study. Diabetologia 2020, 63, 1500–1515. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, D.; Cheng, B.; Chen, J.; Peng, A.; Yang, C.; Liu, C.; Xiong, M.; Deng, A.; Zhang, Y.; et al. Clinical characteristics and outcomes of patients with diabetes and COVID-19 in association with glucose-lowering medication. Diabetes Care 2020, 43, 1399–1407. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, Y.M.; Li, H.; Zhang, X.; Lei, F.; Qin, J.J.; Chen, Z.; Deng, K.Q.; Lin, L.; Chen, M.M.; et al. Metformin is associated with higher incidence of acidosis, but not mortality, in individuals with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 2020, 32, 537–547. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneto, H.; Kimura, T.; Obata, A.; Shimoda, M.; Kaku, K. Multifaceted Mechanisms of Action of Metformin Which Have Been Unraveled One after Another in the Long History. Int. J. Mol. Sci. 2021, 22, 2596. https://doi.org/10.3390/ijms22052596
Kaneto H, Kimura T, Obata A, Shimoda M, Kaku K. Multifaceted Mechanisms of Action of Metformin Which Have Been Unraveled One after Another in the Long History. International Journal of Molecular Sciences. 2021; 22(5):2596. https://doi.org/10.3390/ijms22052596
Chicago/Turabian StyleKaneto, Hideaki, Tomohiko Kimura, Atsushi Obata, Masashi Shimoda, and Kohei Kaku. 2021. "Multifaceted Mechanisms of Action of Metformin Which Have Been Unraveled One after Another in the Long History" International Journal of Molecular Sciences 22, no. 5: 2596. https://doi.org/10.3390/ijms22052596
APA StyleKaneto, H., Kimura, T., Obata, A., Shimoda, M., & Kaku, K. (2021). Multifaceted Mechanisms of Action of Metformin Which Have Been Unraveled One after Another in the Long History. International Journal of Molecular Sciences, 22(5), 2596. https://doi.org/10.3390/ijms22052596