
Sperm Methylome Profiling Can Discern Fertility Levels in the Porcine Biomedical Model

 , , , ,
, , , ,  ,
, 
Abstract
:1. Introduction
2. Results
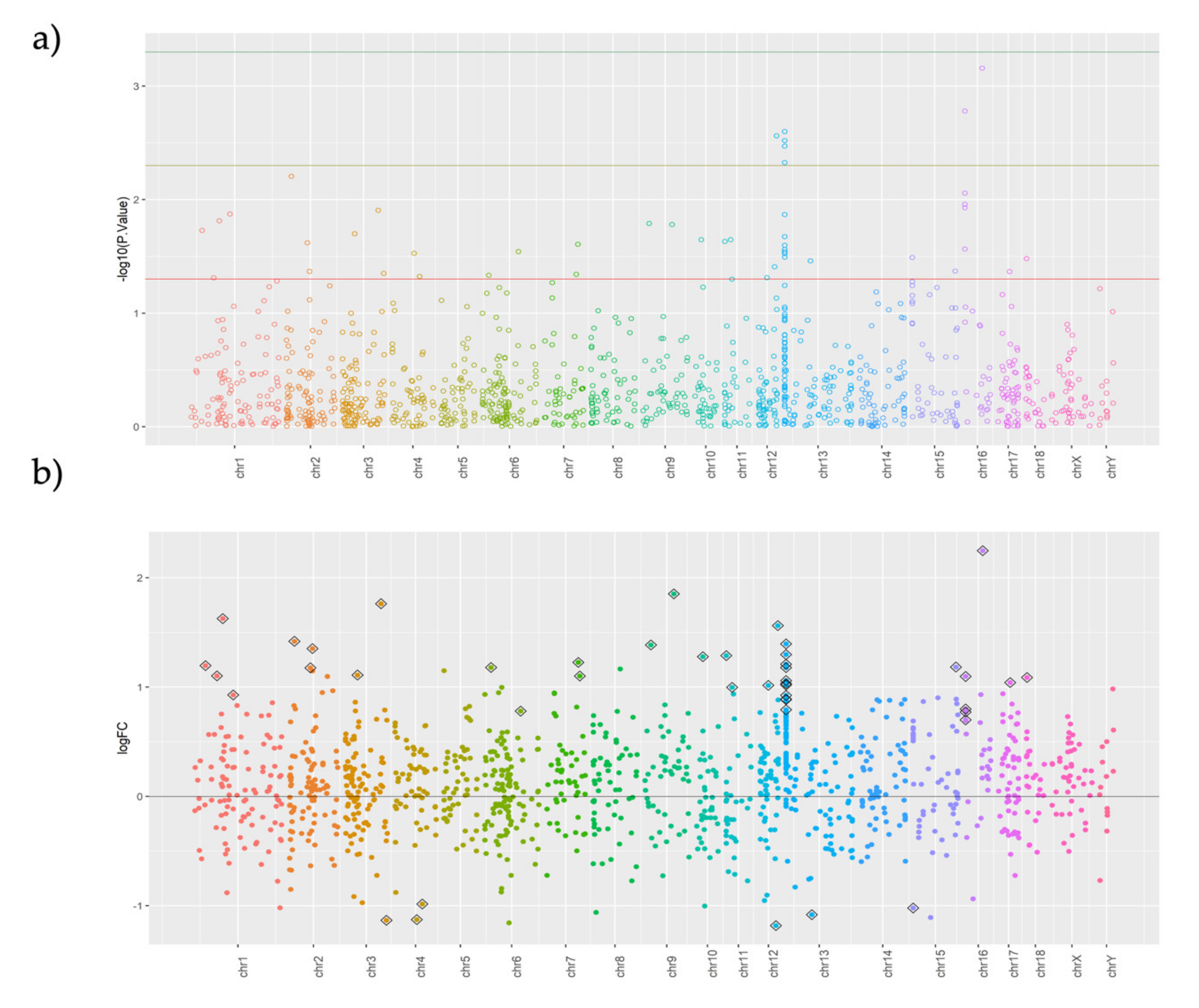
2.1. Identification of SNPs
2.2. Genome-Wide Association Analysis (GWAS) for Boar Fertility
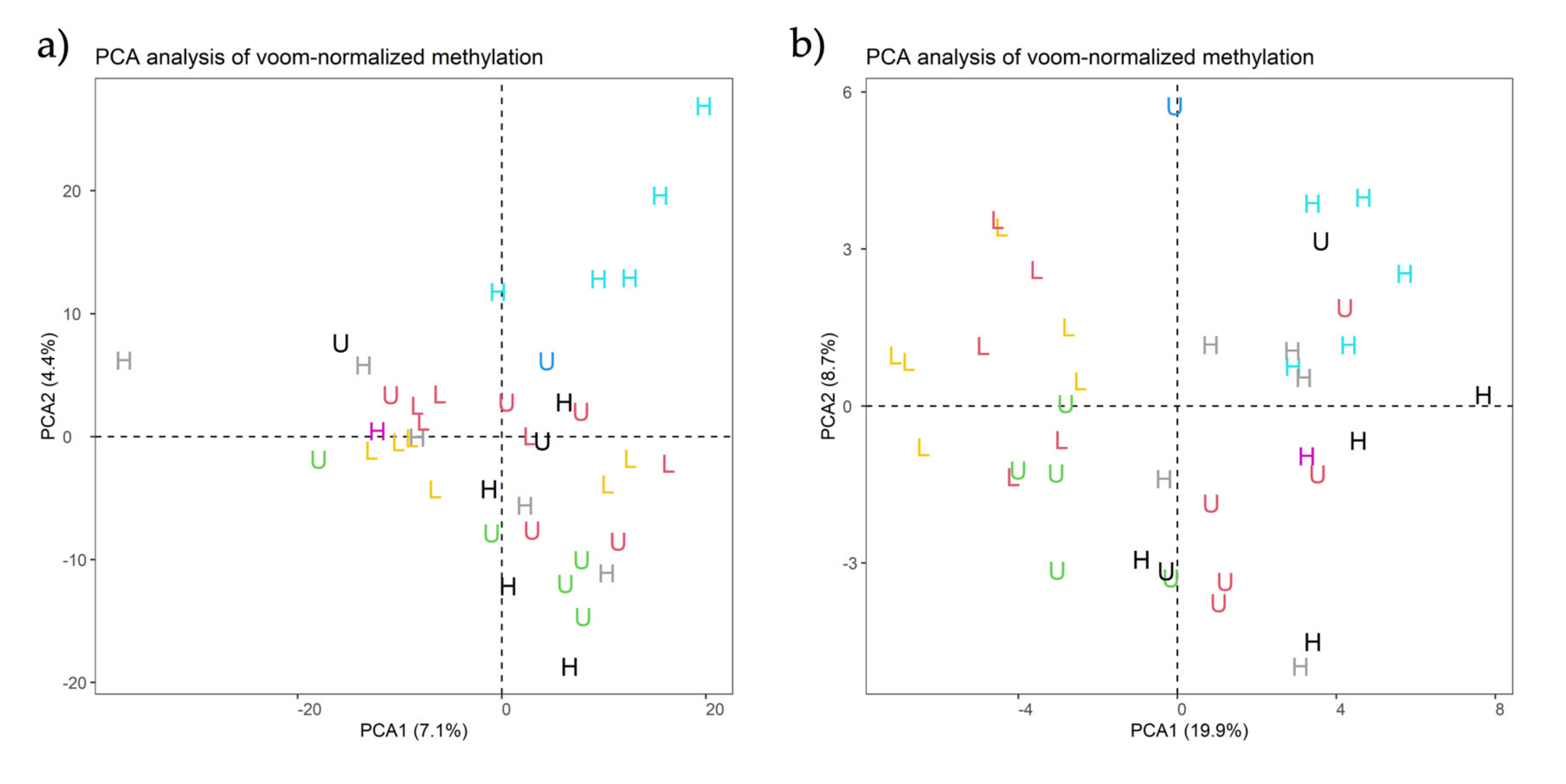
2.3. Putative DMR Identification: Does Fertility Vary with the Season of Semen Collection?
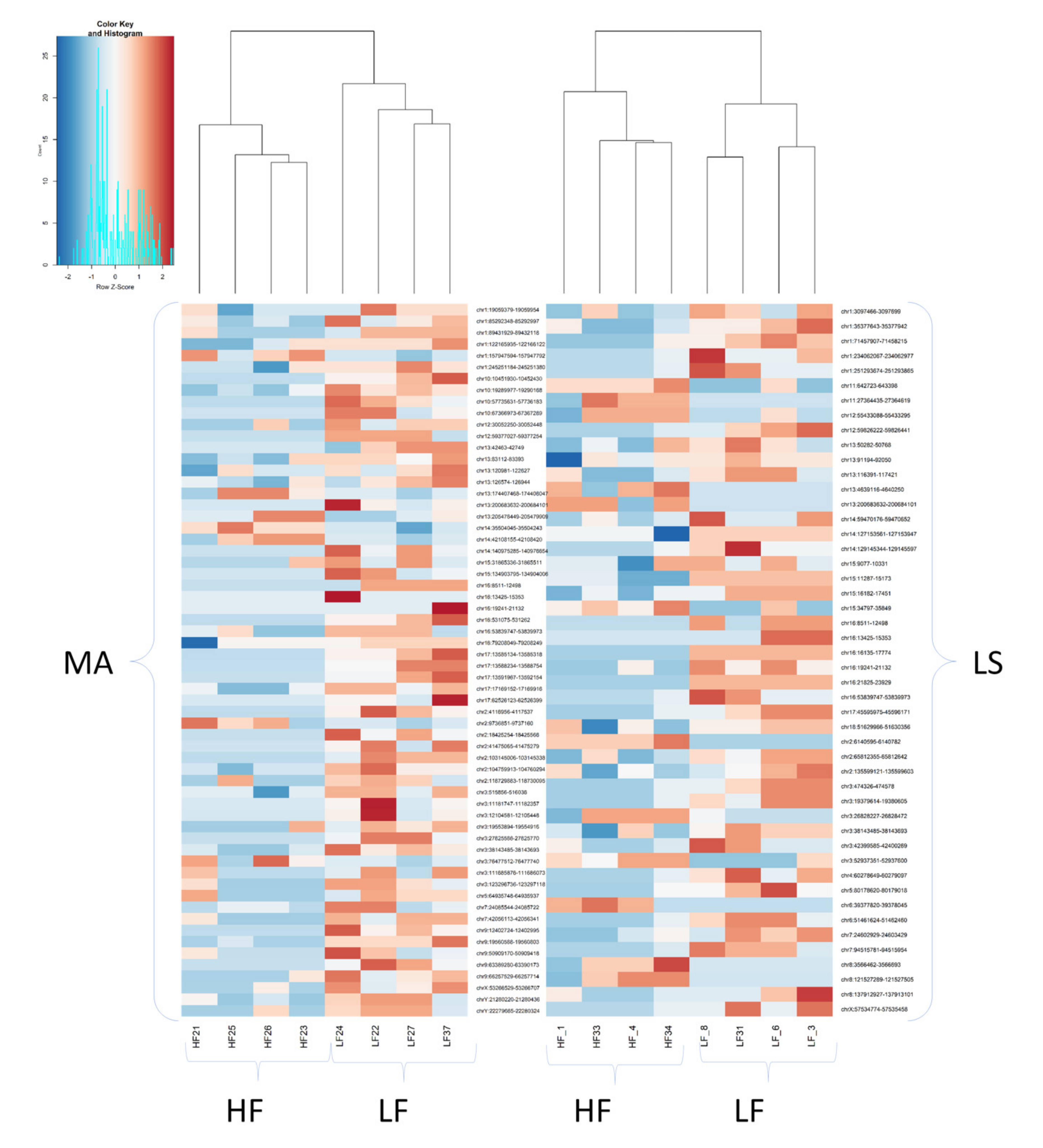
2.4. The Largest Differences between Highly Fertile (HF) and Low Fertile (LF) Boars Are Present in Few Chromosomes
2.5. Most DMRs Are Hypermethylated in Low Fertility Boars
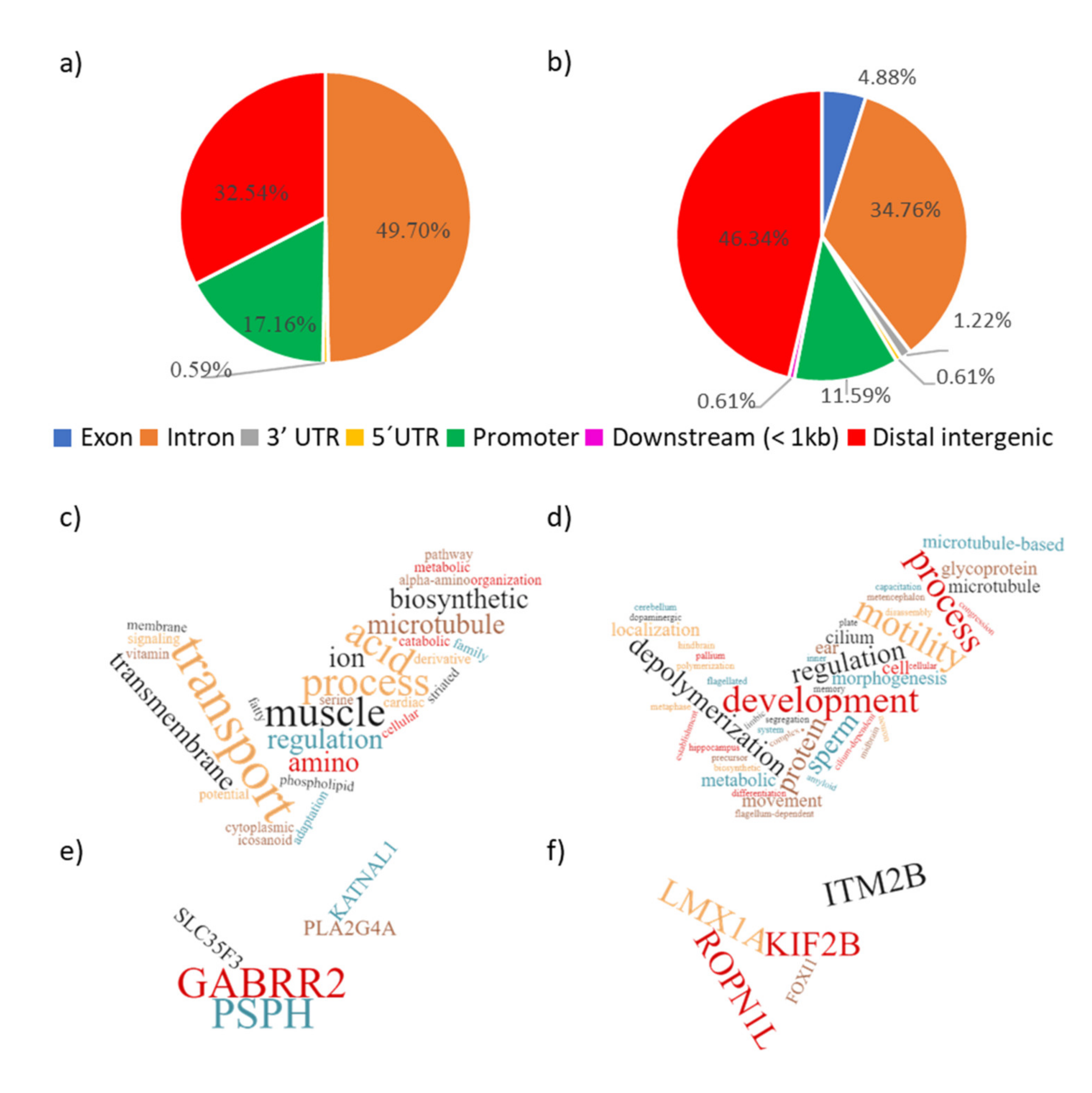
2.6. Genomic Annotation of the SNPs and DMRs
2.7. Overlaps between SNPs and DMRs
3. Discussion
4. Materials and Methods
4.1. Animals and Semen Collection, Evaluation, and Handling
4.2. Experimental Design
4.3. Processing of the Spermatozoa for DNA Isolation
4.4. Preparation of Sequencing Libraries
4.5. Bioinformatics
4.6. Sequencing and Alignment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liffner, S.; Pehrson, I.; García-Calvo, L.; Nedstrand, E.; Zalavary, S.; Hammar, M.; Rodríguez-Martínez, H.; Álvarez-Rodríguez, M. Diagnostics of DNA fragmentation in human spermatozoa: Are sperm chromatin structure analysis and sperm chromatin dispersion tests (SCD-HaloSpermG2 ®) comparable? Andrologia 2019, 51. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Majzoub, A.; Baskaran, S.; Selvam, M.K.P.; Cho, C.L.; Henkel, R.; Finelli, R.; Leisegang, K.; Sengupta, P.; Barbarosie, C.; et al. Sperm DNA fragmentation: A new guideline for clinicians. World J. Mens Health 2020, 38. [Google Scholar] [CrossRef]
- Gudeloglu, A.; Brahmbhatt, J.V.; Parekattil, S.J. Medical management of male infertility in the absence of a specific etiology. Semin. Reprod. Med. 2014, 32, 313–318. [Google Scholar] [CrossRef]
- Walters, E.M.; Wells, K.D.; Bryda, E.C.; Schommer, S.; Prather, R.S. Swine models, genomic tools and services to enhance our understanding of human health and diseases. Lab Anim. 2017, 46, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martínez, H.; Kvist, U.; Saravia, F.; Wallgren, M.; Johannisson, A.; Sanz, L.; Peña, F.J.; Martínez, E.A.; Roca, J.; Vázquez, J.M.; et al. The physiological roles of the boar ejaculate. Soc. Reprod. Fertil. Suppl. 2009, 66, 1–21. [Google Scholar]
- Rodríguez-Martínez, H.; Kvist, U.; Ernerudh, J.; Sanz, L.; Calvete, J.J. Seminal plasma proteins: What role do they play? Am. J. Reprod. Immunol. 2011, 66, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Patiño, C.; Parrilla, I.; Barranco, I.; Vergara-Barberán, M.; Simó-Alfonso, E.F.; Herrero-Martínez, J.M.; Rodriguez-Martínez, H.; Martínez, E.A.; Roca, J. New In-Depth Analytical Approach of the Porcine Seminal Plasma Proteome Reveals Potential Fertility Biomarkers. J. Proteome Res. 2018, 17, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, C.A.; Cambra, J.M.; Parrilla, I.; Roca, J.; Ferreira-Dias, G.; Pallares, F.J.; Lucas, X.; Vazquez, J.M.; Martinez, E.A.; Gil, M.A.; et al. Seminal Plasma Modifies the Transcriptional Pattern of the Endometrium and Advances Embryo Development in Pigs. Front. Vet. Sci. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Rodriguez, M.; Martinez, C.; Wright, D.; Barranco, I.; Roca, J.; Rodriguez-Martinez, H. The Transcriptome of Pig Spermatozoa, and Its Role in Fertility. Int. J. Mol. Sci. 2020, 21, 1572. [Google Scholar] [CrossRef] [Green Version]
- Broekhuijse, M.L.W.J.; Feitsma, H.; Gadella, B.M. Field data analysis of boar semen quality. Reprod. Domest. Anim. 2011, 46, 59–63. [Google Scholar] [CrossRef]
- Roca, J.; Broekhuijse, M.L.W.J.; Parrilla, I.; Rodriguez-Martinez, H.; Martinez, E.A.; Bolarin, A. Boar Differences In Artificial Insemination Outcomes: Can They Be Minimized? Reprod. Domest. Anim. 2015, 50, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foxcroft, G. Application of advanced AI technologies to improve the competitiveness of the pork industry. Int. Pig. 2010, 2010, 25–29. [Google Scholar]
- Rodriguez-Martinez, H. Semen evaluation and handling: Emerging techniques and future development. In Animal Andrology: Theories and Applications; CABI: Wallingford, UK, 2014; pp. 509–549. ISBN 9781780643168. [Google Scholar]
- Rodriguez-Martinez, H. Semen evaluation techniques and their relationship with fertility. Anim. Reprod. 2013, 46, 148–159. [Google Scholar]
- Kumaresan, A.; Das Gupta, M.; Datta, T.K.; Morrell, J.M. Sperm DNA Integrity and Male Fertility in Farm Animals: A Review. Front. Vet. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Krzastek, S.C.; Smith, R.P.; Kovac, J.R. Future diagnostics in male infertility: Genomics, epigenetics, metabolomics and proteomics. Transl. Androl. Urol. 2020, 9, S195–S205. [Google Scholar] [CrossRef] [PubMed]
- Broekhuijse, M.L.W.J.; Šoštarić, E.; Feitsma, H.; Gadella, B.M. The value of microscopic semen motility assessment at collection for a commercial artificial insemination center, a retrospective study on factors explaining variation in pig fertility. Theriogenology 2012, 77. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.L.W.J.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimarães, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Chen, Y.; Liu, X.; Zhao, Y.; Zhu, L.; Liu, A.; Jiang, W.; Peng, X.; Zhang, C.; Tang, Z.; et al. Weighted single-step GWAS identified candidate genes associated with semen traits in a Duroc boar population. BMC Genom. 2019, 20, 797. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, N.; Li, X.; El-Ashram, S.; Wang, Z.; Zhu, L.; Jiang, W.; Peng, X.; Zhang, C.; Chen, Y.; et al. Identifying candidate genes associated with sperm morphology abnormalities using weighted single-step GWAS in a Duroc boar population. Theriogenology 2020, 141, 9–15. [Google Scholar] [CrossRef]
- Khezri, A.; Narud, B.; Stenseth, E.B.; Johannisson, A.; Myromslien, F.D.; Gaustad, A.H.; Wilson, R.C.; Lyle, R.; Morrell, J.M.; Kommisrud, E.; et al. DNA methylation patterns vary in boar sperm cells with different levels of DNA fragmentation. BMC Genom. 2019, 20, 897. [Google Scholar] [CrossRef]
- Pollard, C.A.; Jenkins, T.G. Epigenetic mechanisms within the sperm epigenome and their diagnostic potential. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101481. [Google Scholar] [CrossRef] [PubMed]
- Salilew-Wondim, D.; Gebremedhn, S.; Hoelker, M.; Tholen, E.; Hailay, T.; Tesfaye, D. The role of micrornas in mammalian fertility: From gametogenesis to embryo implantation. Int. J. Mol. Sci. 2020, 21, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, E.; Vavouri, T. Sperm epigenomics: Challenges and opportunities. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Blanco Rodríguez, J.; Camprubí Sánchez, C. Epigenetic transgenerational inheritance. In Advances in Experimental Medicine and Biology; Springer LLC.: New York, NY, USA, 2019; Volume 1166, pp. 57–74. [Google Scholar]
- Pértille, F.; Guerrero-Bosagna, C.; Da Silva, V.H.; Boschiero, C.; Nunes, J.D.R.D.S.; Ledur, M.C.; Jensen, P.; Coutinho, L.L. High-throughput and Cost-effective Chicken Genotyping Using Next-Generation Sequencing. Sci. Rep. 2016, 6, 26929. [Google Scholar] [CrossRef] [PubMed]
- Pértille, F.; Brantsæter, M.; Nordgreen, J.; Coutinho, L.L.; Janczak, A.M.; Jensen, P.; Guerrero-Bosagna, C. DNA methylation profiles in red blood cells of adult hens correlate with their rearing conditions. J. Exp. Biol. 2017, 220, 3579–3587. [Google Scholar] [CrossRef] [Green Version]
- Pértille, F.; Ibelli, A.M.G.; El Sharif, M.; Poleti, M.D.; Fröhlich, A.S.; Rezaei, S.; Ledur, M.C.; Jensen, P.; Guerrero-Bosagna, C.; Coutinho, L.L. Putative Epigenetic Biomarkers of Stress in Red Blood Cells of Chickens Reared Across Different Biomes. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Guerrero-Bosagna, C.; Jensen, P. Optimized method for methylated DNA immuno-precipitation. MethodsX 2015, 2, e432–e439. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.F.; Schnabel, R.D.; Sutovsky, P. Identification of genomic variants causing sperm abnormalities and reduced male fertility. Anim. Reprod. Sci. 2018, 194, 57–62. [Google Scholar] [CrossRef]
- Pausch, H.; Kölle, S.; Wurmser, C.; Schwarzenbacher, H.; Emmerling, R.; Jansen, S.; Trottmann, M.; Fuerst, C.; Götz, K.U.; Fries, R. A Nonsense Mutation in TMEM95 Encoding a Nondescript Transmembrane Protein Causes Idiopathic Male Subfertility in Cattle. PLoS Genet. 2014, 10, e1004044. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.G.; Noonan, E.; Von Eckardstein, S.; Auger, J.; Gordon Baker, H.W.; Behre, H.M.; Haugen, T.B.; Kruger, T.; Wang, C.; Mbizvo, M.T.; et al. World Health Organization reference values for human semen characteristics. Hum. Reprod. Update 2010, 16, 231–245. [Google Scholar] [CrossRef]
- Wang, C.; Swerdloff, R.S. Limitations of semen analysis as a test of male fertility and anticipated needs from newer tests. Fertil. Steril. 2014, 102, 1502–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosova, G.; Scott, N.M.; Niederberger, C.; Prins, G.S.; Ober, C. Genome-Wide association study identifies candidate genes for male fertility traits in humans. Am. J. Hum. Genet. 2012, 90, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Ast, G. Chromatin density and splicing destiny: On the cross-talk between chromatin structure and splicing. EMBO J. 2010, 29, 1629–1636. [Google Scholar] [CrossRef] [Green Version]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Stratil, A.; Peelman, L.J.; Mattheeuws, M.; Van Poucke, M.; Reiner, G.; Geldermann, H. A novel porcine gene, α-1-antichymotrypsin 2 (SERPINA3-2): Sequence, genomic organization, polymorphism and mapping. Gene 2002, 292, 113–119. [Google Scholar] [CrossRef]
- Raffa, R.B.; Connelly, C.D. Supraspinal antinociception produced by [D-Met2]-FMRFamide in mice. Neuropeptides 1992, 22, 195–203. [Google Scholar] [CrossRef]
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Argenti, L.E.; Parmeggiani, B.S.; Leipnitz, G.; Weber, A.; Pereira, G.R.; Bustamante-Filho, I.C. Effects of season on boar semen parameters and antioxidant enzymes in the south subtropical region in Brazil. Andrologia 2018, 50. [Google Scholar] [CrossRef]
- Gòdia, M.; Estill, M.; Castelló, A.; Balasch, S.; Rodríguez-Gil, J.E.; Krawetz, S.A.; Sánchez, A.; Clop, A. A RNA-seq analysis to describe the boar sperm transcriptome and its seasonal changes. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Sancho, S.; Pinart, E.; Briz, M.; Garcia-Gil, N.; Badia, E.; Bassols, J.; Kádár, E.; Pruneda, A.; Bussalleu, E.; Yeste, M.; et al. Semen quality of postpubertal boars during increasing and decreasing natural photoperiods. Theriogenology 2004, 62, 1271–1282. [Google Scholar] [CrossRef]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yang, C.R.; Wei, Y.P.; Zhao, Z.A.; Hou, Y.; Schatten, H.; Sun, Q.Y. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc. Natl. Acad. Sci. USA 2014, 111, 1873–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congras, A.; Yerle-Bouissou, M.; Pinton, A.; Vignoles, F.; Liaubet, L.; Ferchaud, S.; Acloque, H. Sperm DNA methylation analysis in swine reveals conserved and species-specific methylation patterns and highlights an altered methylation at the GNAS locus in infertile boars. Biol. Reprod. 2014, 91, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, P.; Braunschweig, M. DNA methylation patterns at the IGF2-H19 locus in sperm of Swiss Landrace and Swiss Large White boars. J. Anim. Breed. Genet. 2009, 126, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.S.; Yeo, S.; Park, J.S.; Koo, D.B.; Chang, W.K.; Lee, K.K.; Kang, Y.K. DNA methylation state is preserved in the sperm-derived pronucleus of the pig zygote. Int. J. Dev. Biol. 2007, 51, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Gioia, L.; Barboni, B.; Turriani, M.; Capacchietti, G.; Pistilli, M.G.; Berardinelli, P.; Mattioli, M. The capability of reprogramming the male chromatin after fertilization is dependent on the quality of oocyte maturation. Reproduction 2005, 130, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, S.E.; Dudiki, T.; Vijayaraghavan, S.; Carr, D.W. Loss of R2D2 proteins ROPN1 and ROPN1L causes defects in murine sperm motility, phosphorylation, and fibrous sheath integrity. Biol. Reprod. 2013, 88. [Google Scholar] [CrossRef]
- Allbee, A.W.; Rincon-Limas, D.E.; Biteau, B. Lmx1a is required for the development of the ovarian stem cell niche in Drosophila. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomqvist, S.R.; Vidarsson, H.; Söder, O.; Enerbäck, S. Epididymal expression of the forkhead transcription factor Foxi1 is required for male fertility. EMBO J. 2006, 25, 4131–4141. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Huang, Q.; Yuan, S.; Liu, H.; Shi, L.; Yap, Y.T.; Li, W.; Zhen, J.; Zhang, L.; et al. Murine germ cell-specific disruption of Ift172 causes defects in spermiogenesis and male fertility. Reproduction 2020, 159, 409–421. [Google Scholar] [CrossRef]
- Hu, J.R.; Liu, M.; Hou, C.C.; She, Z.Y.; Wang, D.H.; Hao, S.L.; Zhang, Y.P.; Yang, W.X. Gene expression pattern of KIFC3 during spermatogenesis of the skink Eumeces chinensis. Gene 2015, 556, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.E.; Mitchison, T.J.; Desai, A. XKCM1: A Xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell 1996, 84, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Miyata, H.; Castaneda, J.M.; Fujihara, Y.; Yu, Z.; Archambeault, D.R.; Isotani, A.; Kiyozumi, D.; Kriseman, M.L.; Mashiko, D.; Matsumura, T.; et al. Genome engineering uncovers 54 evolutionarily conserved and testis-enriched genes that are not required for male fertility in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 7704–7710. [Google Scholar] [CrossRef] [Green Version]
- Ran, M.; Li, Z.; Cao, R.; Weng, B.; Peng, F.; He, C.; Chen, B. miR-26a suppresses autophagy in swine Sertoli cells by targeting ULK2. Reprod. Domest. Anim. 2018, 53, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Fan, Y.; Wei, L.; He, X.; Yang, J.; Zheng, X. Ablation of TMEM126B protects against oxygen-glucose deprivation/reoxygenation-induced injuries of PC12 cells via maintaining mitochondrial anti-apoptotic functions. Arch. Biochem. Biophys. 2020, 696, 108634. [Google Scholar] [CrossRef] [PubMed]
- Borowska, A.; Szwaczkowski, T.; Kamiński, S.; Hering, D.M.; Kordan, W.; Lecewicz, M. Identification of genome regions determining semen quality in Holstein-Friesian bulls using information theory. Anim. Reprod. Sci. 2018, 192, 206–215. [Google Scholar] [CrossRef]
- Griffin, J.N.; del Viso, F.; Duncan, A.R.; Robson, A.; Hwang, W.; Kulkarni, S.; Liu, K.J.; Khokha, M.K. RAPGEF5 Regulates Nuclear Translocation of β-Catenin. Dev. Cell 2018, 44, 248–260.e4. [Google Scholar] [CrossRef] [Green Version]
- Vadnais, M.L.; Aghajanian, H.K.; Lin, A.; Gerton, G.L. Signaling in sperm: Toward a molecular understanding of the acquisition of sperm motility in the mouse epididymis. Biol. Reprod. 2013, 89. [Google Scholar] [CrossRef] [Green Version]
- Verardo, L.L.; Sevón-Aimonen, M.L.; Serenius, T.; Hietakangas, V.; Uimari, P. Whole-Genome association analysis of pork meat pH revealed three significant regions and several potential genes in Finnish Yorkshire pigs. BMC Genet. 2017, 18, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, M.; Yu, R.; Liu, B.; Tian, Z.; Liu, S. FSCB phosphorylation regulates mouse spermatozoa capacitation through suppressing SUMOylation of ROPN1/ROPN1L. Am. J. Transl. Res. 2016, 8, 2776–2782. [Google Scholar] [PubMed]
- Frayne, J.; Hall, L. A re-evaluation of sperm protein 17 (Sp17) indicates a regulatory role in an A-kinase anchoring protein complex, rather than a unique role in sperm-zona pellucida binding. Reproduction 2002, 124, 767–774. [Google Scholar] [CrossRef]
- Anway, M.D.; Ravindranath, N.; Dym, M.; Griswold, M.D. Identification of a murine testis complementary DNA encoding a homolog to human A-Kinase anchoring protein-associated sperm protein. Biol. Reprod. 2002, 66, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Park, I.; Lee, B.; Jin, S.; Choi, H.; Kwon, J.T.; Kwon, Y.I.; Kim, D.H.; Park, Z.Y.; Cho, C. Identification of heat shock protein 5, calnexin and integral membrane protein 2B as Adam7-interacting membrane proteins in mouse sperm. J. Cell. Physiol. 2011, 226, 1186–1195. [Google Scholar] [CrossRef]
- Choi, H.; Han, C.; Jin, S.; Kwon, J.T.; Kim, J.; Jeong, J.; Kim, J.; Ham, S.; Jeon, S.; Yoo, Y.J.; et al. Reduced Fertility and Altered Epididymal and Sperm Integrity in Mice Lacking ADAM71. Biol. Reprod. 2015, 93, 70. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, X.; Wang, Z.; Li, J.; Xu, Z.; Miao, M.; Chen, G.; Lei, X.; Wu, J.; Shi, H.; et al. MicroRNA expression profile analysis in sperm reveals hsa-mir-191 as an auspicious omen of in vitro fertilization. BMC Genom. 2020, 21, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, G.; Vallée, S.; Rybakin, V.; McGuire, M.V.; Ampudia, J.; Brockmeyer, C.; Salek, M.; Fallen, P.R.; Hoerter, J.A.H.; Munshi, A.; et al. Themis controls thymocyte selection through regulation of T cell antigen receptor-mediated signaling. Nat. Immunol. 2009, 10, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Montiel, W.; Martínez-Núñez, M.A.; Ramón-Ugalde, J.P.; Román-Ponce, S.I.; Calderón-Chagoya, R.; Zamora-Bustillos, R. Genome-wide association study reveals candidate genes for litter size traits in pelibuey sheep. Animals 2020, 10, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Wang, K.; Yang, Q.; Zhou, J.; Chen, D.; Liu, Y.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; et al. Whole-genome re-sequencing association study for direct genetic effects and social genetic effects of six growth traits in Large White pigs. Sci. Rep. 2019, 9, 9667. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.K.; Lee, J.B.; Ramayo-Caldas, Y.; Kim, S.D.; Cho, E.S.; Kim, Y.S.; Cho, K.H.; Lee, D.H.; Park, H.B. Single-step genome-wide association study for social genetic effects and direct genetic effects on growth in Landrace pigs. Sci. Rep. 2020, 10, 14958. [Google Scholar] [CrossRef]
- Smith, L.B.; Milne, L.; Nelson, N.; Eddie, S.; Brown, P.; Atanassova, N.; O’Bryan, M.K.; O’Donnell, L.; Rhodes, D.; Wells, S.; et al. KATNAL1 regulation of sertoli cell microtubule dynamics is essential for spermiogenesis and male fertility. PLoS Genet. 2012, 8, e1002697. [Google Scholar] [CrossRef] [Green Version]
- Fedick, A.M.; Eckert, K.; Thompson, K.; Forman, E.J.; Devkota, B.; Treff, N.R.; Taylor, D.; Scott, R.T. Lack of association of KATNAL1 gene sequence variants and azoospermia in humans. J. Assist. Reprod. Genet. 2014, 31, 1065–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, K.; Roychoudhury, S.; Bora, S.; Dehury, B.; Parida, P.; Das, S.; Das, R.; Dohutia, C.; Nath, S.; Deb, B.; et al. Molecular Modeling and Dynamics Simulation Analysis of KATNAL1 for Identification of Novel Inhibitor of Sperm Maturation. Comb. Chem. High Throughput Screen. 2017, 20, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Adeola, A.C.; Xie, H.B.; Zhang, Y.P. Genomic and transcriptomic analyses reveal selection of genes for puberty in Bama Xiang pigs. Zool. Res. 2018, 39, 424–430. [Google Scholar] [PubMed]
- Zhang, X.; Wang, C.; Zhang, Y.; Ju, Z.; Qi, C.; Wang, X.; Huang, J.; Zhang, S.; Li, J.; Zhong, J.; et al. Association between an alternative promoter polymorphism and sperm deformity rate is due to modulation of the expression of KATNAL1 transcripts in Chinese Holstein bulls. Anim. Genet. 2014, 45, 641–651. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and genotyping loci de novo from short-read sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team, R. R: A Language and Environment for Statistical Computing; R Foundation Statistical Computing: Vienna, Austria, 2011; Volume 1, p. 409. [Google Scholar]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [Green Version]
- Niazi, U.; Geyer, K.K.; Vickers, M.J.; Hoffmann, K.F.; Swain, M.T. DISMISS: Detection of stranded methylation in MeDIP-Seq data. BMC Bioinform. 2016, 17, 295. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, R.G.; Qin, T.; Sartor, M.A. Novel bioinformatics methods for toxicoepigenetics. In Toxicoepigenetics: Core Principles and Applications; Elsevier: Amsterdam, The Netherlands, 2018; pp. 265–288. ISBN 9780128124338. [Google Scholar]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Chavez, L.; Jozefczuk, J.; Grimm, C.; Dietrich, J.; Timmermann, B.; Lehrach, H.; Herwig, R.; Adjaye, J. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010, 20, 1441–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Down, T.A.; Rakyan, V.K.; Turner, D.J.; Flicek, P.; Li, H.; Kulesha, E.; Gräf, S.; Johnson, N.; Herrero, J.; Tomazou, E.M.; et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat. Biotechnol. 2008, 26, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin/Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIP seeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contrast | Group1 | Group2 | Purpose of Contrast |
|---|---|---|---|
| HvL | HF | LF | to compare genomic data between HF and LF individuals |
| H_LSvsMA | H_LS | H_MA | to compare the effect of the sampling date between late-summer (LS) and mid-autumn (MA) on HF individuals |
| L_LSvsMA | L_LS | L_MA | to compare the effect of the sampling date between late-summer (LS) and mid-autumn (MA) on LF individuals |
| HvsL_LS | H_LS | L_LS | to compare genomic data between HF and LF individuals at the LS sampling period |
| HvsL_MA | H_MA | L_MA | to compare genomic data between HF and LF individuals at the MA sampling period |
| Location | Site Summary in the Analyzed Population n = 7 | Annotation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref | Alt | A1 | F_A1 HF | F_A 1 LF | A2 | SeqCpG | Annotation | Distance to TSS | ENSEMBL ID | GENE ID | Description | |
| chr1:13745053 | G | A | A | 0.5 | 0.25 | G | GC | Intron | 28,711 | ENSSSCG00000004081 | ||
| chr1:13745059 | T | C | C | 0.5 | 0.25 | T | TC | 28,717 | ||||
| chr1:13745092 | C | T | T | 0.5 | 0.25 | C | CG | 28,750 | ||||
| chr1:54055492 | A | G | G | 0 | 0.625 | A | AG | Distal Intergenic | 33,321 | ENSSSCG00000035731 | − | |
| chr1:54055493 | G | C | C | 0 | 0.625 | G | GC | 33,320 | ||||
| chr1:54055625 | G | C | C | 0 | 0.625 | G | GA | 33,188 | ||||
| chr1:57269839 | T | A | A | 0 | 0.625 | T | TG | Intron | −3712 | ENSSSCG00000024249 | GABRR2 | gamma-aminobutyric acid type A receptor subunit rho2 [Source:NCBI gene (formerly Entrezgene);Acc:100522289] |
| chr1:57269858 | T | C | C | 0 | 0.625 | T | TC | −3731 | ||||
| chr1:57277143 | C | A | A | 0 | 0.625 | C | CA | −11,016 | ||||
| chr1:57528770 | G | A | A | 0 | 0.625 | G | GC | Intron | 61,949 | ENSSSCG00000050825 | ||
| chr1:57621352 | G | A | A | 0 | 0.625 | G | GC | Promoter | 2902 | ENSSSCG00000004322 | ANKRD6 | ankyrin repeat domain 6 [Source:NCBI gene (formerly Entrezgene);Acc:102167335] |
| chr1:57621367 | G | A | A | 0 | 0.625 | G | GG | 2917 | ||||
| chr1:57621370 | T | C | C | 0 | 0.625 | T | TT | 2920 | ||||
| chr1:128707723 | C | T | T | 0 | 0.625 | C | TG | Promoter | 1229 | ENSSSCG00000022039 | ||
| chr1:140846691 | A | C | C | 0.5 | 0.25 | A | AT | Intron | 170,343 | ENSSSCG00000038547 | GABRB3 | gamma-aminobutyric acid type A receptor subunit beta3 [Source:VGNC Symbol;Acc:VGNC:88308] |
| chr1:167452422 | C | G | G | 0 | 0.625 | C | GA | Distal Intergenic | 63,364 | ENSSSCG00000045133 | ||
| chr1:168947778 | T | G | G | 0 | 0.625 | T | TG | Intron | −147,253 | ENSSSCG00000045715 | ||
| chr2:10168387 | C | T | C | 0 | 0.625 | T | TC | Intron | 3002 | ENSSSCG00000013087 | TKFC | triokinase and FMN cyclase [Source:NCBI gene (formerly Entrezgene);Acc:100520121] |
| chr2:10168389 | C | G | G | 0 | 0.625 | C | CA | 3000 | ||||
| chr2:13387750 | G | T | T | 0 | 0.625 | G | TG | Promoter | −2942 | ENSSSCG00000031163 | smoothelin like 1 [Source:NCBI gene (formerly Entrezgene);Acc:110259259] | |
| chr2:13387762 | G | A | A | 0 | 0.625 | G | AA | −2954 | ||||
| chr3:16873170 | G | C | C | 0.5 | 0.25 | G | GT | Distal Intergenic | −4753 | ENSSSCG00000007748 | PSPH | phosphoserine phosphatase [Source:VGNC Symbol;Acc:VGNC:91931] |
| chr6:3925771 | C | G | G | 0 | 0.625 | C | CA | Intron | 18,808 | ENSSSCG00000002669 | CRISPLD2 | cysteine rich secretory protein LCCL domain containing 2 [Source:VGNC Symbol;Acc:VGNC:96958] |
| chr6:3925776 | C | G | G | 0 | 0.625 | C | CC | 18,803 | ||||
| chr6:3925779 | C | T | T | 0 | 0.625 | C | CC | 18,800 | ||||
| chr6:7702343 | G | C | C | 0 | 0.625 | G | GG | Intron | −4397 | ENSSSCG00000045637 | ||
| chr6:7702375 | G | A | A | 0 | 0.625 | G | GG | −4429 | ||||
| chr6:101129470 | A | G | G | 0 | 0.625 | A | AT | Distal Intergenic | 35,288 | ENSSSCG00000049298 | ||
| chr7:87106399 | G | T | T | 0.5 | 0.25 | G | GA | Distal Intergenic | −3090 | ENSSSCG00000002263 | SLCO3A1 | solute carrier organic anion transporter family member 3A1 [Source:VGNC Symbol;Acc:VGNC:93197] |
| chr7:115966284 | C | T | T | 0 | 0.625 | C | CG | Intron | 5808 | ENSSSCG00000030371 | SERPINA3-2 | alpha-1-antichymotrypsin 2 [Source:NCBI gene (formerly Entrezgene);Acc:396686] |
| chr7:115966307 | C | T | T | 0 | 0.625 | C | CC | 5785 | ||||
| chr7:115982628 | C | A | A | 0 | 0.625 | C | AA | 4776 | ||||
| chr7:115982655 | G | T | T | 0 | 0.625 | G | TG | 4749 | ||||
| chr8:1318578 | C | T | T | 0.5 | 0.25 | C | CG | Intron | 5424 | ENSSSCG00000034633 | CFAP99 | cilia and flagella associated protein 99 [Source:VGNC Symbol;Acc:VGNC:86608] |
| chr8:1328667 | A | G | G | 0.5 | 0.25 | A | AG | 15,513 | ||||
| chr9:40003949 | A | G | G | 0.5 | 0.25 | A | GC | Distal Intergenic | −46,046 | ENSSSCG00000050685 | ||
| chr9:40003978 | T | C | C | 0.5 | 0.25 | T | TG | −46,017 | ||||
| chr9:45947327 | G | T | T | 0.5 | 0.25 | G | TT | Intron | 15,089 | ENSSSCG00000028204 | TREH | trehalase [Source:VGNC Symbol;Acc:VGNC:94382] |
| chr11:7034734 | A | T | T | 0.5 | 0.25 | A | AA | Intron | 50,584 | ENSSSCG00000009326 | KATNAL1 | katanin catalytic subunit A1 like 1 [Source:VGNC Symbol;Acc:VGNC:89310] |
| chr11:16127229 | G | A | A | 0.5 | 0.25 | G | GG | Intron | 67,905 | ENSSSCG00000040538 | WDFY2 | WD repeat and FYVE domain containing 2 [Source:VGNC Symbol;Acc:VGNC:94902] |
| chr11:61861680 | G | C | C | 0 | 0.625 | G | GG | Intron | 323,962 | ENSSSCG00000048281 | ||
| chr12:19677123 | G | A | A | 0 | 0.625 | G | GC | Intron | −4345 | ENSSSCG00000040316 | U2 | U2 spliceosomal RNA [Source:RFAM;Acc:RF00004] |
| chr12:41648810 | G | A | A | 0.5 | 0.25 | G | GT | Intron | −78,192 | ENSSSCG00000040177 | ASIC2 | acid sensing ion channel subunit 2 [Source:VGNC Symbol;Acc:VGNC:97893] |
| chr12:41648838 | T | C | C | 0.5 | 0.25 | T | TC | −78,164 | ||||
| chr12:42246850 | A | G | G | 0 | 0.625 | A | AA | Intron | −61,012 | ENSSSCG00000050001 | ||
| chr14:4991961 | T | G | G | 1 | 0.125 | T | TT | Distal Intergenic | 419,328 | ENSSSCG00000018795 | ||
| chr14:24479770 | A | G | G | 0 | 0.625 | A | GT | Intron | 46,536 | ENSSSCG00000009748 | RIMBP2 | RIMS binding protein 2 [Source:VGNC Symbol;Acc:VGNC:92306] |
| chr14:56252347 | G | C | C | 0.5 | 0.25 | G | GG | Promoter | −1423 | ENSSSCG00000043646 | ||
| chr15:63579255 | C | T | T | 0.5 | 0.25 | C | CC | Distal Intergenic | −13,443 | ENSSSCG00000015872 | GPD2 | glycerol-3-phosphate dehydrogenase 2 [Source:VGNC Symbol;Acc:VGNC:96330] |
| chr15:63579295 | A | G | G | 0.5 | 0.25 | A | AA | −13,403 | ||||
| chr15:140048049 | C | T | T | 0.5 | 0.25 | C | CG | Intron | −12,312 | ENSSSCG00000016368 | FARP2 | FERM, ARH/RhoGEF and pleckstrin domain protein 2 [Source:VGNC Symbol;Acc:VGNC:95809] |
| chr15:140089430 | A | T | T | 0.5 | 0.25 | A | AA | 14,636 | ||||
| chr16:53406651 | T | C | C | 0 | 0.625 | T | CC | Intron | −81,737 | ENSSSCG00000017003 | KCNIP1 | potassium voltage-gated channel interacting protein 1 [Source:VGNC Symbol;Acc:VGNC:89348] |
| chrY:5970972 | T | A | T | 1 | 0 | A | AA | Intron | 24,002 | ENSSSCG00000039894 | ||
| chrY:5970979 | G | C | G | 1 | 0 | C | CG | 24,009 | ||||
| Contrasts | Number of DMRs (p ≤ 0.05) | Number of DMRs (FDR ≤ 0.5) |
|---|---|---|
| HF–LF | 46 | 0 |
| LS–LA | 40 | 0 |
| MA–LA | 41 | 0 |
| MA–LS | 49 | 0 |
| HF_LS–HF_MA | 87 | 7 |
| LF_LS–LF_MA | 27 | 0 |
| HF_LS–LF_LS | 48 | 4 |
| HF_MA–LF_MA | 62 | 3 |
| Location | logFC | p-Value | adj.p.Val | Annotation | Distance ToTSS | EMSEMBL ID | GENE ID | Description | CG |
|---|---|---|---|---|---|---|---|---|---|
| chr1:35377643-35377942 | 1.20 | 1.86 × 10−2 | 9.96 × 10−1 | Intron | 44,178 | ENSSSCG00000024392 | THEMIS | thymocyte selection associated (Source:VGNC Symbol;Acc:VGNC:98369) | 9 |
| chr1:71457907-71458215 | 1.10 | 4.88 × 10−2 | 9.96 × 10−1 | Promoter | −502 | ENSSSCG00000045756 | 8 | ||
| chr1:89431929-89432118 | 1.63 | 1.54 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −167,999 | ENSSSCG00000046698 | 1 | ||
| chr1:122165935-122166122 | 0.93 | 1.34 × 10−2 | 9.96 × 10−1 | Intron | 26,493 | ENSSSCG00000004648 | FAM227B | family with sequence similarity 227 member B (Source:VGNC Symbol;Acc:VGNC:87961) | 8 |
| chr2:15503913-15504126 | 1.42 | 6.22 × 10−3 | 9.40 × 10−1 | Intron | 27,523 | ENSSSCG00000020542 | U6 | U6 spliceosomal RNA (Source:RFAM;Acc:RF00026) | 10 |
| chr2:65812355-65812642 | 1.18 | 2.39 × 10−2 | 9.96 × 10−1 | Intron | 90,547 | ENSSSCG00000013754 | calcium voltage-gated channel subunit alpha1 A (Source:VGNC Symbol;Acc:VGNC:99705) | 6 | |
| chr2:72272984-72273185 | 1.35 | 4.28 × 10−2 | 9.96 × 10−1 | 5’ UTR | −3761 | ENSSSCG00000013556 | adhesion G protein-coupled receptor E1 (Source:VGNC Symbol;Acc:VGNC:85124) | 21 | |
| chr3:38143485-38143693 | 1.11 | 1.99 × 10−2 | 9.96 × 10−1 | Intron | −29,370 | ENSSSCG00000007950 | ADCY9 | adenylate cyclase 9 (Source:VGNC Symbol;Acc:VGNC:85113) | 6 |
| chr3:111685876-111686073 | 1.76 | 1.24 × 10−2 | 9.96 × 10−1 | Promoter | 2167 | ENSSSCG00000026367 | IFT172 | intraflagellar transport 172 (Source:VGNC Symbol;Acc:VGNC:89046) | 2 |
| chr3:128777380-128777600 | −1.13 | 4.46 × 10−2 | 9.96 × 10−1 | Intron | 8370 | ENSSSCG00000008646 | RNF144A | ring finger protein 144A (Source:VGNC Symbol;Acc:VGNC:92358) | 2 |
| chr4:68193523-68193723 | −1.13 | 2.97 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 31,282 | ENSSSCG00000023848 | VXN | vexin (Source:VGNC Symbol;Acc:VGNC:94889) | 4 |
| chr4:85493823-85493995 | −0.98 | 4.74 × 10−2 | 9.96 × 10−1 | Intron | 86,294 | ENSSSCG00000006329 | LMX1A | LIM homeobox transcription factor 1 alpha (Source:VGNC Symbol;Acc:VGNC:89770) | 6 |
| chr6:19702050-19702740 | 1.18 | 4.64 × 10−2 | 9.96 × 10−1 | Intron | −5977 | ENSSSCG00000002812 | KIFC3 | kinesin family member C3 (Source:VGNC Symbol;Acc:VGNC:89479) | 8 |
| chr6:112264267-112264451 | 0.78 | 2.87 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 259,145 | ENSSSCG00000042906 | 8 | ||
| chr7:99786455-99786655 | 1.23 | 4.55 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −57,879 | ENSSSCG00000051045 | 1 | ||
| chr7:104533019-104533586 | 1.10 | 2.47 × 10−2 | 9.96 × 10−1 | Promoter | −284 | ENSSSCG00000049060 | 16 | ||
| chr9:19560588-19560803 | 1.39 | 1.62 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 12,942 | ENSSSCG00000014905 | TMEM126B | transmembrane protein 126B (Source:NCBI gene (formerly Entrezgene);Acc:100626990) | 6 |
| chr9:91128754-91129011 | 1.85 | 1.65 × 10−2 | 9.96 × 10−1 | Intron | 70,639 | ENSSSCG00000015383 | RAPGEF5 | Rap guanine nucleotide exchange factor 5 (Source:VGNC Symbol;Acc:VGNC:92094) | 1 |
| chr10:19289977-19290168 | 1.28 | 2.26 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −16,228 | ENSSSCG00000033907 | 5 | ||
| chr11:1065989-1066196 | 1.29 | 2.33 × 10−2 | 9.96 × 10−1 | 3’ UTR | 28,854 | ENSSSCG00000009276 | XPO4 | exportin 4 (Source:VGNC Symbol;Acc:VGNC:95004) | 1 |
| chr11:1065989-1066196 | 1.29 | 2.33 × 10−2 | 9.96 × 10−1 | 3’ UTR | 28,854 | 1 | |||
| chr11:19350117-19350833 | 1.00 | 2.26 × 10−2 | 9.96 × 10−1 | Intron | 5261 | ENSSSCG00000009403 | ITM2B | integral membrane protein 2B (Source:NCBI gene (formerly Entrezgene);Acc:595120) | 12 |
| chr12:30052250-30052448 | 1.02 | 4.87 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −109,077 | ENSSSCG00000017599 | KIF2B | kinesin family member 2B (Source:VGNC Symbol;Acc:VGNC:89468) | 3 |
| chr12:53660655-53660898 | −1.18 | 3.90 × 10−2 | 9.96 × 10−1 | Exon | −50,792 | ENSSSCG00000039032 | U6 | U6 spliceosomal RNA (Source:RFAM;Acc:RF00026) | 16 |
| chr12:59826222-59826441 | 1.56 | 2.74 × 10−3 | 6.80 × 10−1 | Intron | 19,267 | ENSSSCG00000018045 | ULK2 | unc-51 like autophagy activating kinase 2 (Source:VGNC Symbol;Acc:VGNC:94695) | 6 |
| chr13:7508-7838 | 1.18 | 3.37 × 10−3 | 6.80 × 10−1 | Distal Intergenic | 281,632 | ENSSSCG00000046931 | 9 | ||
| chr13:34107-34423 | 0.89 | 2.52 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 255,047 | 5 | |||
| chr13:37450-37723 | 1.03 | 2.98 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 251,747 | 5 | |||
| chr13:42463-42749 | 1.40 | 4.72 × 10−3 | 8.14 × 10−1 | Distal Intergenic | 246,721 | 7 | |||
| chr13:83112-83393 | 1.21 | 1.35 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 206077 | 5 | |||
| chr13:96003-97046 | 1.06 | 3.01 × 10−3 | 6.80 × 10−1 | Distal Intergenic | 192,424 | 18 | |||
| chr13:104471-105251 | 0.89 | 2.12 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 184,219 | 12 | |||
| chr13:109921-111269 | 0.79 | 3.21 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 178,201 | 27 | |||
| chr13:120981-122627 | 0.92 | 2.74 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 166,843 | 34 | |||
| chr13:133660-134567 | 1.30 | 2.51 × 10−3 | 6.80 × 10−1 | Distal Intergenic | 154,903 | 22 | |||
| chr13:138579-139179 | 1.02 | 2.88 × 10−2 | 9.96 × 10−1 | Distal Intergenic | 150,291 | 10 | |||
| chr13:80744108-80744758 | −1.08 | 3.46 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −44,636 | ENSSSCG00000011666 | CLSTN2 | calsyntenin 2 (Source:VGNC Symbol;Acc:VGNC:86785) | 6 |
| chr15:34797-35849 | −1.02 | 3.24 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −4170 | ENSSSCG00000047217 | 23 | ||
| chr15:134010175-134010376 | 1.18 | 4.26 × 10−2 | 9.96 × 10−1 | Intron | −4041 | ENSSSCG00000049095 | 5 | ||
| chr16:8511-12498 | 1.10 | 1.66 × 10−3 | 6.80 × 10−1 | Distal Intergenic | −30,855 | ENSSSCG00000022306 | ROPN1L | rhophilin associated tail protein 1 like (Source:VGNC Symbol;Acc:VGNC:92406) | 98 |
| chr16:13425-15353 | 0.77 | 1.10 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −28,000 | 45 | |||
| chr16:16135-17774 | 0.70 | 2.72 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −25,579 | 41 | |||
| chr16:19241-21132 | 0.80 | 8.75 × 10−3 | 9.96 × 10−1 | Distal Intergenic | −22,221 | 44 | |||
| chr16:21825-23929 | 0.77 | 1.18 × 10−2 | 9.96 × 10−1 | Distal Intergenic | −19,424 | ENSSSCG00000022306 | ROPN1L | rhophilin associated tail protein 1 like (Source:VGNC Symbol;Acc:VGNC:92406) | 50 |
| chr16:53839747-53839973 | 2.25 | 6.94 × 10−4 | 6.80 × 10−1 | Promoter | −638 | ENSSSCG00000017009 | FOXI1 | forkhead box I1 (Source:VGNC Symbol;Acc:VGNC:88208) | 14 |
| chr17:36220445-36220684 | 1.04 | 4.29 × 10−2 | 9.96 × 10−1 | Intron | −19,300 | ENSSSCG00000007249 | NOL4L | COMM domain containing 7 (Source:NCBI gene (formerly Entrezgene);Acc:100514995) | 7 |
| chr17:36220445-36220684 | 1.04 | 4.29 × 10−2 | 9.96 × 10−1 | Intron | −19,300 | 7 | |||
| chr18:1373929-1374129 | 1.09 | 3.31 × 10−2 | 9.96 × 10−1 | Intron | 12,247 | ENSSSCG00000037841 | ssc-mir-153 | ssc-mir-153 (Source:miRBase;Acc:MI0002454) | 7 |
| Samples | Treat. | Sample Number | Depth ± SD | Number of bp Sequenced ± SD | Breadth (bp Sequenced/Depth) ± SD | % of the % of Susscr11.1 Covered ± SD | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GBS | All | 11 | 33.81 | ± | 13.38 | 2746210261 | ± | 1651232585 | 72531345 | ± | 24303149 | 2.93 | ± | 0.98 |
| Hi | 4 | 34.92 | ± | 15.21 | 2979989907 | ± | 1926403257 | 75972199 | ± | 28862451 | 3.07 | ± | 1.16 | |
| Low | 3 | 35.98 | ± | 11.79 | 2875589615 | ± | 1539581798 | 75762826 | ± | 19130954 | 3.06 | ± | 0.77 | |
| UF | 4 | 31.09 | ± | 16.00 | 2415396101 | ± | 1887272897 | 66666880 | ± | 28630213 | 2.69 | ± | 1.15 | |
| GBS + MEDIP | All | 39 | 19.30 | ± | 2.35 | 128051103 | ± | 35431948 | 6721573 | ± | 1830176 | 0.27 | ± | 0.07 |
| Hi | 15 | 19.72 | ± | 2.79 | 136199403 | ± | 49129755 | 7021683 | ± | 2391520 | 0.28 | ± | 0.10 | |
| Low | 11 | 19.07 | ± | 1.38 | 117088079 | ± | 24901730 | 6219760 | ± | 1640964 | 0.25 | ± | 0.07 | |
| UF | 13 | 19.01 | ± | 2.55 | 127925623 | ± | 21202711 | 6799904 | ± | 1157695 | 0.27 | ± | 0.05 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pértille, F.; Alvarez-Rodriguez, M.; da Silva, A.N.; Barranco, I.; Roca, J.; Guerrero-Bosagna, C.; Rodriguez-Martinez, H. Sperm Methylome Profiling Can Discern Fertility Levels in the Porcine Biomedical Model. Int. J. Mol. Sci. 2021, 22, 2679. https://doi.org/10.3390/ijms22052679
Pértille F, Alvarez-Rodriguez M, da Silva AN, Barranco I, Roca J, Guerrero-Bosagna C, Rodriguez-Martinez H. Sperm Methylome Profiling Can Discern Fertility Levels in the Porcine Biomedical Model. International Journal of Molecular Sciences. 2021; 22(5):2679. https://doi.org/10.3390/ijms22052679
Chicago/Turabian StylePértille, Fabio, Manuel Alvarez-Rodriguez, Arthur Nery da Silva, Isabel Barranco, Jordi Roca, Carlos Guerrero-Bosagna, and Heriberto Rodriguez-Martinez. 2021. "Sperm Methylome Profiling Can Discern Fertility Levels in the Porcine Biomedical Model" International Journal of Molecular Sciences 22, no. 5: 2679. https://doi.org/10.3390/ijms22052679