
Manipulation of the Glass Transition Properties of a High-Solid System Made of Acrylic Acid-N,N′-Methylenebisacrylamide Copolymer Grafted on Hydroxypropyl Methyl Cellulose


Abstract
:1. Introduction
2. Results and Discussion
2.1. Physicochemical Characterization of the HPMC-AAc System
2.2. Morphological Characteristics of the HPMC-AAc Matrix
2.3. Application of Mechanical Spectroscopy on the HPMC-AAc Network
2.4. Calorimetric Glass Transition Temperature of the HPMC-AAc System
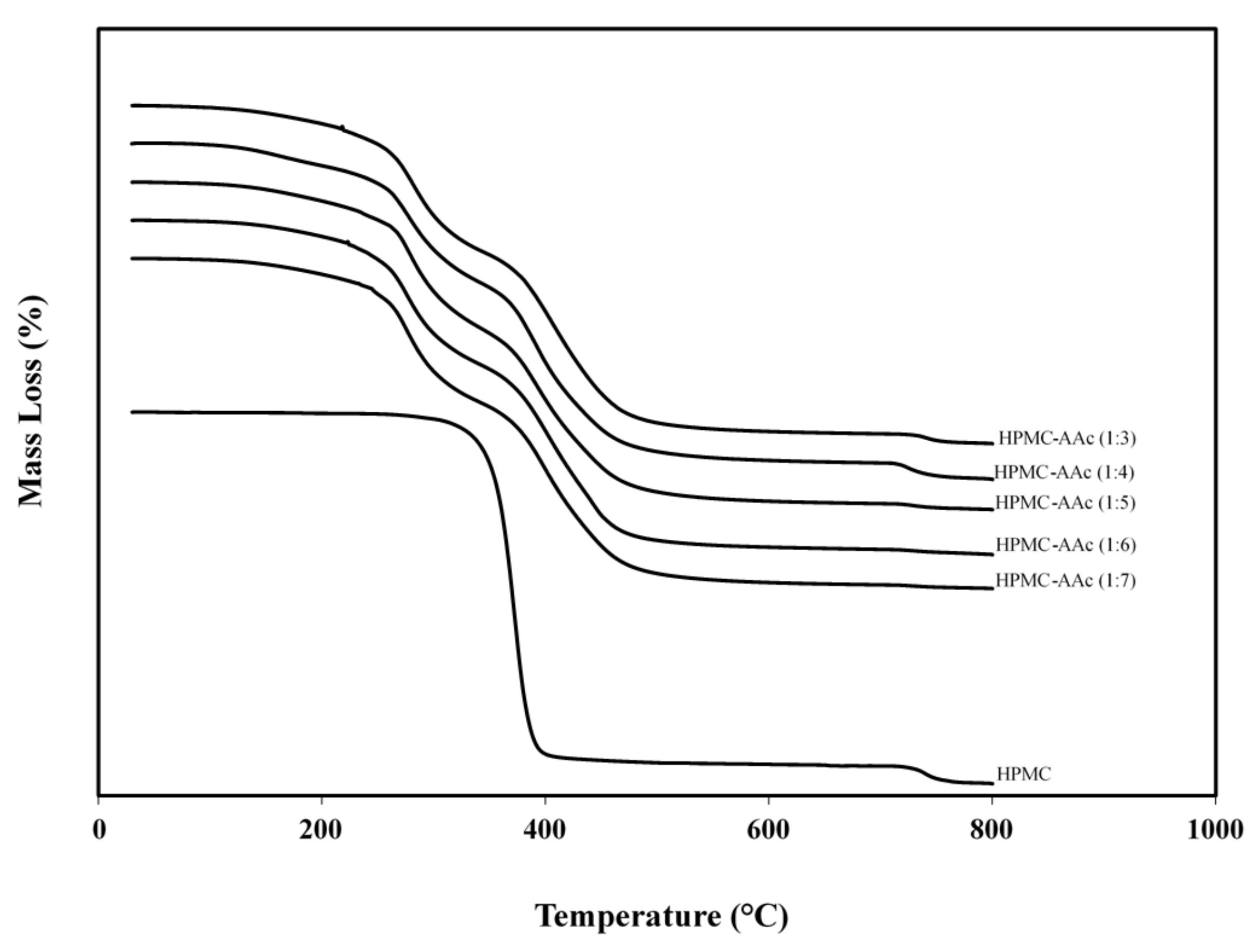
2.5. Thermogravimetric Analysis of the High-Solid HPMC-AAc Matrix
3. Materiala and Methods
3.1. Materials
3.2. Sample Preparation
3.3. Experimental Analysis
3.3.1. Fourier Transform Infrared Spectroscopy (FTIR)
3.3.2. X-Ray Diffraction Analysis (XRD)
3.3.3. Scanning Electron Microscopy (SEM)
3.3.4. Rheological Measurements
3.3.5. Modulated Scanning Calorimetry (MDSC)
3.3.6. Thermogravimetric Analysis (TGA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, E.M.; Aggor, F.S.; Awad, A.M.; El-Aref, A.T. An innovative method for preparation of nanometal hydroxide superabsorbent hydrogel. Carbohydr. Polym. 2013, 91, 693–698. [Google Scholar] [CrossRef]
- Akhtar, M.F.; Hanif, M.; Ranjha, N.M. Methods of synthesis of hydrogels A review. Saudi Pharm. J. 2016, 24, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, D.; Salomé Veiga, A.; Castanho, M.A.R.B. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedron, C.N.; Torres, M.D.; da Silva Lima, J.A.; Silva, P.I.; Silva, F.D.; Oliveira, V.X. Novel designed VmCT1 analogs with increased antimicrobial activity. Eur. J. Med. Chem. 2017, 126, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Woodruff, T.M.; Clark, R.J.; Martin, D.J.; Minchin, R.F. Release of bioactive peptides from polyurethane films in vitro and in vivo: Effect of polymer composition. Acta Biomater. 2016, 41, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huichao, W.; Shouying, D.; Yang, L.; Ying, L.; Di, W.J. The application of biomedical polymer material hydroxy propyl methyl cellulose(HPMC) in pharmaceutical preparations. J. Chem. Pharm. Res. 2014, 6, 155–160. [Google Scholar]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Ma, G. Microencapsulation of protein drugs for drug delivery: Strategy, preparation, and applications. J. Controlled Release 2014, 193, 324–340. [Google Scholar] [CrossRef]
- Yao, R.S.; Zhang, W.B.; Yang, X.Z.; Liu, J.; Liu, H.T. HPMC/PAA hybrid nanogels via aqueous-phase synthesis for controlled delivery of insulin. Biomater. Sci. 2014, 2, 1761–1767. [Google Scholar] [CrossRef]
- Shoaib, M.H.; Siddiqi, S.A.; Yousuf, R.I.; Zaheer, K.; Hanif, M.; Rehana, S.; Jabeen, S. Development and evaluation of hydrophilic colloid matrix of famotidine tablets. AAPS PharmSciTech 2010, 11, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penhasi, A. Microencapsulation of probiotic bacteria using thermo-sensitive sol-gel polymers for powdered infant formula. J. Microencapsul. 2015, 32, 372–380. [Google Scholar] [CrossRef]
- Zhang, H.; Zhai, Y.; Wang, J.; Zhai, G. New progress and prospects: The application of nanogel in drug delivery. Mater. Sci. Eng. C 2016, 60, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Malhotra, S.; Molina, M.; Haag, R. Micro- and nanogels with labile crosslinks-from synthesis to biomedical applications. Chem. Soc. Rev. 2015, 44, 1948–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichenbaum, G.M.; Kiser, P.F.; Dobrynin, A.V.; Simon, S.A.; Needham, D. Investigation of the swelling response and loading of ionic microgels with drugs and proteins: The dependence on cross-link density. Macromolecules 1999, 32, 4867–4878. [Google Scholar] [CrossRef]
- Mijovic, J.; Shen, M.; Sy, J.W.; Mondragon, I. Dynamics and morphology in nanostructured thermoset network/block copolymer blends during network formation. Macromolecules 2000, 33, 5235–5244. [Google Scholar] [CrossRef]
- Pikabea, A.; Aguirre, G.; Miranda, J.I.; Ramos, J.; Forcada, J. Understanding of nanogels swelling behavior through a deep insight into their morphology. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2017–2025. [Google Scholar] [CrossRef]
- Qiao, Z.Y.; Zhang, R.; Du, F.S.; Liang, D.H.; Li, Z.C. Multi-responsive nanogels containing motifs of ortho ester, oligo(ethylene glycol) and disulfide linkage as carriers of hydrophobic anti-cancer drugs. J. Control. Release 2011, 152, 57–66. [Google Scholar] [CrossRef]
- Soni, G.; Yadav, K.S. Nanogels as potential nanomedicine carrier for treatment of cancer: A mini review of the state of the art. Saudi Pharm. J. 2016, 24, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Soni, K.S.; Desale, S.S.; Bronich, T.K. Nanogels: An overview of properties, biomedical applications and obstacles to clinical translation. J. Control. Release 2016, 240, 109–126. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, S.I.; Kajiki, M.; Kuriyama, H.; Kobayashi, T.R. Complete regression of xenografted human carcinomas by a paclitaxel-carboxymethyl dextran conjugate (AZ10992). J. Control. Release 2007, 117, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.V.; Bronich, T.K.; Kabanov, A.V. Nanosized cationic hydrogels for drug delivery: Preparation, properties and interactions with cells. Adv. Drug Deliv. Rev. 2002, 54, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Söderlind, E.; Viridén, A.; Schug, B.; Abrahamsson, B.; Knopke, C.; Tajarobi, F.; Blume, H.; Anschütz, M.; Welinder, A.; et al. The influence of hydroxypropyl methylcellulose (HPMC) molecular weight, concentration and effect of food on in vivo erosion behavior of HPMC matrix tablets. J. Control. Release 2014, 187, 50–58. [Google Scholar] [CrossRef]
- Miriyala, N.; Ouyang, D.; Perrie, Y.; Lowry, D.; Kirby, D.J. Activated carbon as a carrier for amorphous drug delivery: Effect of drug characteristics and carrier wettability. Eur. J. Pharm. Biopharm. 2017, 115, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.R. Fundamentals of Transport Phenomena in Polymer Membranes. In Comprehensive Membrane Science and Engineering; Elsevier: Amsterdam, The Netherlands, 2010; pp. 75–90. [Google Scholar]
- Martínez, L.M.; Videa, M.; Sosa, N.G.; Ramírez, J.H.; Castro, S. Long-term stability of new co-amorphous drug binary systems: Study of glass transitions as a function of composition and shelf time. Molecules 2016, 21, 1712. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, A.; Sako, K.; Maitani, Y. Influence of manufacturing factors on physical stability and solubility of solid dispersions containing a low glass transition temperature drug. Chem. Pharm. Bull. 2012, 60, 1366–1371. [Google Scholar] [CrossRef] [Green Version]
- Kasapis, S. Definition and applications of the network glass transition temperature. Food Hydrocoll. 2006, 20, 218–228. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Kraisit, P.; Limmatvapirat, S.; Nunthanid, J.; Sriamornsak, P.; Luangtana-Anan, M. Preparation and characterization of hydroxypropyl methylcellulose/polycarbophil mucoadhesive blend films using a mixture design approach. Chem. Pharm. Bull. 2017, 65, 284–294. [Google Scholar] [CrossRef] [Green Version]
- The, D.P.; Debeaufort, F.; Luu, D.; Voilley, A. Moisture barrier, wetting and mechanical properties of shellac/agar or shellac/cassava starch bilayer bio-membrane for food applications. J. Membr. Sci. 2008, 325, 277–283. [Google Scholar]
- Barik, A.; Patnaik, T.; Parhi, P.; Swain, S.K.; Dey, R.K. Synthesis and characterization of new shellac–hydroxypropylmethylcellulose composite for pharmaceutical applications. Polym. Bull. 2017, 74, 3467–3485. [Google Scholar] [CrossRef]
- Kasapis, S. Glass transition phenomena in dehydrated model systems and foods: A review. Dry. Technol. 2005, 23, 731–757. [Google Scholar] [CrossRef]
- Kasapis, S.; Sablani, S.S. A fundamental approach for the estimation of the mechanical glass transition temperature in gelatin. Int. J. Biol. Macromol. 2005, 36, 71–78. [Google Scholar] [CrossRef]
- Kasapis, S. Recent advances and future challenges in the explanation and exploitation of the network glass transition of high sugar/biopolymer mixtures. Crit. Rev. Food Sci. Nutr. 2008, 48, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Kasapis, S.; Al-Marhoobi, I.M.; Mitchell, J.R. Testing the validity of comparisons between the rheological and the calorimetric glass transition temperatures. Carbohydr. Res. 2003, 338, 787–794. [Google Scholar] [CrossRef]
- Lomellini, P. Williams-Landel-Ferry versus Arrhenius behaviour: Polystyrene melt viscoelasticity revised. Polymer 1992, 33, 4983–4989. [Google Scholar] [CrossRef]
- Chaudhary, V.; Small, D.M.; Kasapis, S. Effect of a glassy gellan/polydextrose matrix on the activity of α-d-glucosidase. Carbohydr. Polym. 2013, 95, 389–396. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: Hoboken, NJ, USA, 1980. [Google Scholar]
- White, R.P.; Lipson, J.E.G. Polymer Free Volume and Its Connection to the Glass Transition. Macromolecules 2016, 49, 3987–4007. [Google Scholar] [CrossRef]
- Van der Put, T.A.C.M. Theoretical derivation of the WLF- and annealing equations. J. Non-Cryst. Solids 2010, 356, 394–399. [Google Scholar] [CrossRef]
- Yao, R.; Xu, J.; Lu, X.; Deng, S. Phase transition behavior of HPMC-AA and preparation of HPMC-PAA nanogels. J. Nanomater. 2011, 2011. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A view of the hydrophobic effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Schmidt, S.J. Water and solids mobility in foods. Adv. Food Nutr. Res. 2004, 48, 1–101. [Google Scholar]
- McNeill, I.C.; Sadeghi, S.M.T. Thermal stability and degradation mechanisms of poly(acrylic acid) and its salts: Part 1-Poly(acrylic acid). Polym. Degrad. Stab. 1990, 29, 233–246. [Google Scholar] [CrossRef]
- Iqbal, F.M.; Ahmad, M.; Rashid, A. Synthesis and in vitro characterisation of hydroxyprpopyl methylcellulose-graft-poly(acrylic acid/2-acrylamido-2-methyl-1-propanesulfonic acid) polymeric network for controlled release of captopril. Acta Pol. Pharm. 2016, 73, 183–196. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPMC:AAc | DSC Tg (°C) | Rheological Tg (°C) | Arrhenius Parameters | WLF Parameters | |||||
|---|---|---|---|---|---|---|---|---|---|
| Tg onset (°C) | Tg midpoint (°C) | Tg endpoint (°C) | Ea (kJ mol−1) | C1 | C2 (deg) | f | αf × 104 (deg−1) | ||
| 1:3 | −2.3 | 13.6 | 22.7 | 40 | 311 | 21.9 | 123.2 | 0.020 | 2.0 |
| 1:4 | −6.1 | 5.6 | 16.5 | 26 | 206 | 15.6 | 93.8 | 0.030 | 3.0 |
| 1:5 | −12.9 | −1.1 | 11.5 | 18 | 268 | 21.8 | 119.3 | 0.020 | 2.0 |
| 1:6 | −18.4 | −8.4 | −4.1 | 12 | 274 | 10.2 | 36.6 | 0.040 | 1.2 |
| 1:7 | −21.5 | −14.2 | −11.7 | −14 | 113 | 12.8 | 39.0 | 0.030 | 9.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassar, N.; Whitehead, F.; Istivan, T.; Shanks, R.; Kasapis, S. Manipulation of the Glass Transition Properties of a High-Solid System Made of Acrylic Acid-N,N′-Methylenebisacrylamide Copolymer Grafted on Hydroxypropyl Methyl Cellulose. Int. J. Mol. Sci. 2021, 22, 2682. https://doi.org/10.3390/ijms22052682
Nassar N, Whitehead F, Istivan T, Shanks R, Kasapis S. Manipulation of the Glass Transition Properties of a High-Solid System Made of Acrylic Acid-N,N′-Methylenebisacrylamide Copolymer Grafted on Hydroxypropyl Methyl Cellulose. International Journal of Molecular Sciences. 2021; 22(5):2682. https://doi.org/10.3390/ijms22052682
Chicago/Turabian StyleNassar, Nazim, Felicity Whitehead, Taghrid Istivan, Robert Shanks, and Stefan Kasapis. 2021. "Manipulation of the Glass Transition Properties of a High-Solid System Made of Acrylic Acid-N,N′-Methylenebisacrylamide Copolymer Grafted on Hydroxypropyl Methyl Cellulose" International Journal of Molecular Sciences 22, no. 5: 2682. https://doi.org/10.3390/ijms22052682