
Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction


Abstract
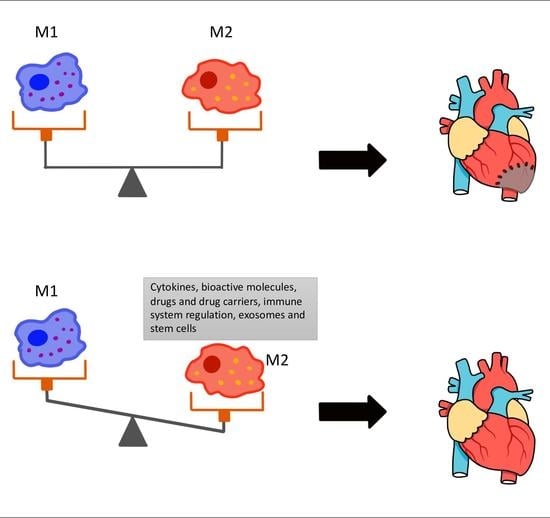
:
1. Introduction
2. Overview of Macrophage Phenotypes and Polarization States
3. The Role of M1 and M2 Macrophages in MI
4. Therapeutic Implications
4.1. Cytokines, Bioactive Molecules and Drugs
4.2. Regulation of the Immune System
4.3. Stem Cells and Exosomes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mensah, G.A.; Roth, G.A.; Fuster, V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J. Am. Coll. Cardiol. 2019, 74, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- McClellan, M.; Brown, N.; Califf, R.M.; Warner, J.J. Call to action: Urgent challenges in cardiovascular disease: A presidential advisory from the American Heart Association. Circulation 2019, 139, e44–e54. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N. Heart disease and stroke statistics—2020 update: A report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association. Cardiovascular Disease: A Costly Burden for America Projections through 2035; American Heart Association: Washington, DC, USA, 2017. [Google Scholar]
- Nowbar, A.N.; Gitto, M.; Howard, J.P.; Francis, D.P.; Al-Lamee, R. Mortality from ischemic heart disease: Analysis of data from the World Health Organization and coronary artery disease risk factors from NCD Risk Factor Collaboration. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005375. [Google Scholar] [CrossRef] [PubMed]
- Vogel, B.; Claessen, B.E.; Arnold, S.V.; Chan, D.; Cohen, D.J.; Giannitsis, E.; Gibson, C.M.; Goto, S.; Katus, H.A.; Kerneis, M.; et al. ST-segment elevation myocardial infarction. Nat. Rev. Dis. Primers 2019, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.; Bagchi, A.K.; Vinayak, K.; Bello-Klein, A.; Singal, P.K. Innate immune response in the pathogenesis of heart failure in survivors of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H435–H445. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Dodsworth, M.; Thum, T. Inflammatory cells and their non-coding RNAs as targets for treating myocardial infarction. Basic Res. Cardiol. 2018, 114, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, R.D.; Chong, J.J.H. The Cardiac Injury Immune Response as a Target for Regenerative and Cellular Therapies. Clin. Ther. 2020, 42, 1923–1943. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Dittrich, A.; Lauridsen, H. Myocardial infarction and the immune response-scarring or regeneration? A comparative look at mammals and popular regenerating animal models. J. Immunol. Regen. Med. 2019, 4, 100016. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Kaffka Genaamd Dengler, S.E.; Odille, C.A.; Mishra, M.; Van der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; De Kleijn, D.P.V.; De Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11, 599511. [Google Scholar] [CrossRef] [PubMed]
- Daseke, M.J., 2nd; Valerio, F.M.; Kalusche, W.J.; Ma, Y.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res. Cardiol. 2019, 114, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saparov, A.; Ogay, V.; Nurgozhin, T.; Chen, W.C.; Mansurov, N.; Issabekova, A.; Zhakupova, J. Role of the immune system in cardiac tissue damage and repair following myocardial infarction. Inflamm. Res. 2017, 66, 739–751. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. J. Lab. Clin. Med. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2-and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef]
- Li, Y.; Li, Q.; Fan, G.C. Macrophage Efferocytosis in Cardiac Pathophysiology and Repair. Shock 2021, 55, 177–188. [Google Scholar] [CrossRef]
- Deniset, J.F.; Belke, D.; Lee, W.Y.; Jorch, S.K.; Deppermann, C.; Hassanabad, A.F.; Turnbull, J.D.; Teng, G.; Rozich, I.; Hudspeth, K.; et al. Gata6 (+) Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity 2019, 51, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Song, L.; Wang, L.; Yukht, A.; Ruther, H.; Li, F.; Qin, M.; Ghiasi, H.; Sharifi, B.G.; Shah, P.K. Deficiency of GATA3-Positive Macrophages Improves Cardiac Function Following Myocardial Infarction or Pressure Overload Hypertrophy. J. Am. Coll. Cardiol. 2018, 72, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, B.G.; Yang, M.; Shah, P.K. Aging and GATA3-positive macrophages. Aging 2019, 11, 2179–2180. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Suto, A.; Suzuki, K.; Kobayashi, Y.; Nakajima, H. Matrix metalloproteinase-12 produced by Ly6C (low) macrophages prolongs the survival after myocardial infarction by preventing neutrophil influx. J. Mol. Cell. Cardiol. 2019, 131, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Shrivastava, R.; Shukla, N. Attributes of alternatively activated (M2) macrophages. Life Sci. 2019, 224, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef]
- Kloc, M.; Ghobrial, R.M.; Wosik, J.; Lewicka, A.; Lewicki, S.; Kubiak, J.Z. Macrophage functions in wound healing. J. Tissue Eng. Regen. Med. 2019, 13, 99–109. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef]
- Frodermann, V.; Nahrendorf, M. Macrophages and Cardiovascular Health. Physiol. Rev. 2018, 98, 2523–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Hirschi, K.K. Tissue-Resident Macrophage Development and Function. Front. Cell Dev. Biol. 2021, 8, 1750. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, A.C.; Soldano, S.; Contini, P.; Tomatis, V.; Ruaro, B.; Paolino, S.; Brizzolara, R.; Montagna, P.; Sulli, A.; Pizzorni, C.; et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir. Res. 2018, 19, 186. [Google Scholar] [CrossRef]
- López-Janeiro, Á.; Padilla-Ansala, C.; De Andrea, C.E.; Hardisson, D.; Melero, I. Prognostic value of macrophage polarization markers in epithelial neoplasms and melanoma. A systematic review and meta-analysis. Mod. Pathol. 2020, 33, 1458–1465. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef]
- Bellamri, N.; Morzadec, C.; Lecureur, V.; Joannes, A.; Wollin, L.; Jouneau, S.; Vernhet, L. Effects of Nintedanib on the M1 and M2a polarization of human macrophages. Eur. Respir. Soc. 2018, 52. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Gerrick, K.Y.; Gerrick, E.R.; Gupta, A.; Wheelan, S.J.; Yegnasubramanian, S.; Jaffee, E.M. Transcriptional profiling identifies novel regulators of macrophage polarization. PLoS ONE 2018, 13, e0208602. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.Y.; Xiong, Y.Y.; Lu, X.T.; Yang, Y.J. Regulation of Type 2 Immunity in Myocardial Infarction. Front. Immunol. 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lin, J.; Lian, X.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Liang, X.; Hao, W. M2a and M2b macrophages predominate in kidney tissues and M2 subpopulations were associated with the severity of disease of IgAN patients. Clin. Immunol. 2019, 205, 8–15. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Kieler, M.; Hofmann, M.; Schabbauer, G. More than just protein building blocks: How amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1 (LPS+) vs. Classically and M2 (LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.X.; Gustafson, H.H.; Jackson, D.L.; Pun, S.H.; Trapnell, C. Trajectory analysis quantifies transcriptional plasticity during macrophage polarization. Sci. Rep. 2020, 10, 12273. [Google Scholar] [CrossRef]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [Green Version]
- Bosco, M.C. Macrophage polarization: Reaching across the aisle? J. Allergy Clin. Immunol. 2019, 143, 1348–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vorst, E.P.C.; Weber, C. Novel Features of Monocytes and Macrophages in Cardiovascular Biology and Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e30–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Hardie, J.; Landis, R.F.; Mas-Rosario, J.A.; Chattopadhyay, A.N.; Keshri, P.; Sun, J.; Rizzo, E.M.; Gopalakrishnan, S.; Farkas, M.E. High-content and high-throughput identification of macrophage polarization phenotypes. Chem. Sci. 2020, 11, 8231–8239. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.L.; Koh, T.J. Macrophage-based therapeutic strategies in regenerative medicine. Adv. Drug Deliv. Rev. 2017, 122, 74–83. [Google Scholar] [CrossRef]
- Chan, M.W.Y.; Viswanathan, S. Recent progress on developing exogenous monocyte/macrophage-based therapies for inflammatory and degenerative diseases. Cytotherapy 2019, 21, 393–415. [Google Scholar] [CrossRef]
- Barman, P.K.; Koh, T.J. Macrophage Dysregulation and Impaired Skin Wound Healing in Diabetes. Front. Cell Dev. Biol. 2020, 8, 528. [Google Scholar] [CrossRef]
- Giannarelli, C.; Fernandez, D.M. Manipulating Macrophage Polarization to Fix the Broken Heart: Challenges and Hopes. J. Am. Coll. Cardiol. 2018, 72, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Koenig, A.L.; Lavine, K.J.; Apte, R.S. Macrophage Plasticity and Function in the Eye and Heart. Trends Immunol. 2019, 40, 825–841. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Li, Y.; Fan, G.C. Tissue-Resident Macrophages in the Control of Infection and Resolution of Inflammation. Shock 2021, 55, 14–23. [Google Scholar] [CrossRef]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, S.A.; Dunne, A.; Monaghan, M.G. The Role of Macrophages in the Infarcted Myocardium: Orchestrators of ECM Remodeling. Front. Cardiovasc. Med. 2019, 6, 101. [Google Scholar] [CrossRef] [Green Version]
- Nicolás-Ávila, J.A.; Hidalgo, A.; Ballesteros, I. Specialized functions of resident macrophages in brain and heart. J. Leukoc. Biol. 2018, 104, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Nahrendorf, M. Novel functions of macrophages in the heart: Insights into electrical conduction, stress, and diastolic dysfunction. Eur. Heart J. 2020, 41, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.; Cabrera-Fuentes, H.A.; Irei, J.; Hausenloy, D.J.; Boisvert, W.A. Role of Macrophages in Cardioprotection. Int. J. Mol. Sci. 2019, 20, 2474. [Google Scholar] [CrossRef] [Green Version]
- Honold, L.; Nahrendorf, M. Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef]
- Liu, S.; Chen, J.; Shi, J.; Zhou, W.; Wang, L.; Fang, W.; Zhong, Y.; Chen, X.; Chen, Y.; Sabri, A.; et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res. Cardiol. 2020, 115, 22. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Mentkowski, K.I.; Euscher, L.M.; Patel, A.; Alevriadou, B.R.; Lang, J.K. Monocyte recruitment and fate specification after myocardial infarction. Am. J. Physiol. Cell Physiol. 2020, 319, C797–C806. [Google Scholar] [CrossRef]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Rivera Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [Green Version]
- Gombozhapova, A.; Rogovskaya, Y.; Shurupov, V.; Rebenkova, M.; Kzhyshkowska, J.; Popov, S.V.; Karpov, R.S.; Ryabov, V. Macrophage activation and polarization in post-infarction cardiac remodeling. J. Biomed. Sci. 2017, 24, 13. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Rong, J. Macrophage Polarization as a Therapeutic Target in Myocardial Infarction. Curr. Drug Targets 2018, 19, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Ito, T.; Fields, L.; Shiraishi, M.; Ichihara, Y.; Sato, N.; Podaru, M.; Kainuma, S.; Tanaka, H.; Suzuki, K. IL-4 as a Repurposed Biological Drug for Myocardial Infarction through Augmentation of Reparative Cardiac Macrophages: Proof-of-Concept Data in Mice. Sci. Rep. 2017, 7, 6877. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Ma, Y.; Iyer, R.P.; DeLeon-Pennell, K.Y.; Yabluchanskiy, A.; Garrett, M.R.; Lindsey, M.L. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res. Cardiol. 2017, 112, 33. [Google Scholar] [CrossRef]
- Jing, R.; Long, T.Y.; Pan, W.; Li, F.; Xie, Q.Y. IL-6 knockout ameliorates myocardial remodeling after myocardial infarction by regulating activation of M2 macrophages and fibroblast cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6283–6291. [Google Scholar] [CrossRef]
- Carlson, S.; Helterline, D.; Asbe, L.; Dupras, S.; Minami, E.; Farris, S.; Stempien-Otero, A. Cardiac macrophages adopt profibrotic/M2 phenotype in infarcted hearts: Role of urokinase plasminogen activator. J. Mol. Cell. Cardiol. 2017, 108, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, Y.; Jin, J.Y.; Li, G.H.; Guo, Y.Z.; Yi, H.Y.; Zhang, J.R.; Lu, Y.J.; Zhang, J.L.; Li, C.Y.; et al. CD226 deletion improves post-infarction healing via modulating macrophage polarization in mice. Theranostics 2020, 10, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Guo, M.; Zhou, P.; Xiao, J.; Ji, X. TSLP promote M2 macrophages polarization and cardiac healing after myocardial infarction. Biochem. Biophys. Res. Commun. 2019, 516, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Shen, X.; Whiteman, M.; Xin, H.; Shen, Y.; Xin, X.; Moore, P.K.; Zhu, Y.Z. Hydrogen Sulfide Mitigates Myocardial Infarction via Promotion of Mitochondrial Biogenesis-Dependent M2 Polarization of Macrophages. Antioxid. Redox Signal. 2016, 25, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Liu, Y.; Li, T.; Tuo, Q. Role of Hydrogen Sulfide in Chronic Diseases. DNA Cell Biol. 2020, 39, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Wang, F.; Pei, S.; Bai, R.; Cong, X.; Nie, Y.; Chen, X. Hydrogen Sulfide Promotes Cardiomyocyte Proliferation and Heart Regeneration via ROS Scavenging. Oxid. Med. Cell. Longev. 2020, 2020, 1412696. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, C.; Wang, X.; He, T.; Li, L.; Liang, X.; Wang, L.; Song, L.; Wei, Y.; Wu, Q.; et al. Hyaluronic Acid Oligosaccharides Improve Myocardial Function Reconstruction and Angiogenesis against Myocardial Infarction by Regulation of Macrophages. Theranostics 2019, 9, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.; McNeill, B.; Podrebarac, J.; Hosoyama, K.; Sedlakova, V.; Cron, G.; Smyth, D.; Seymour, R.; Goel, K.; Liang, W. Injectable human recombinant collagen matrices limit adverse remodeling and improve cardiac function after myocardial infarction. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Luo, D.; Zhao, Y.; Rong, J. N-Propargyl caffeate amide (PACA) prevents cardiac fibrosis in experimental myocardial infarction by promoting pro-resolving macrophage polarization. Aging 2020, 12, 5384–5398. [Google Scholar] [CrossRef] [PubMed]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The Effects of Resveratrol in Patients with Cardiovascular Disease and Heart Failure: A Narrative Review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Du, Y.; Shi, K.; Yang, Y.; Yang, Z. Resveratrol improves cardiac function by promoting M2-like polarization of macrophages in mice with myocardial infarction. Am. J. Transl. Res. 2019, 11, 5212–5226. [Google Scholar] [PubMed]
- Torrieri, G.; Fontana, F.; Figueiredo, P.; Liu, Z.; Ferreira, M.P.A.; Talman, V.; Martins, J.P.; Fusciello, M.; Moslova, K.; Teesalu, T.; et al. Dual-peptide functionalized acetalated dextran-based nanoparticles for sequential targeting of macrophages during myocardial infarction. Nanoscale 2020, 12, 2350–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Mordechai, T.; Kain, D.; Holbova, R.; Landa, N.; Levin, L.P.; Elron-Gross, I.; Glucksam-Galnoy, Y.; Feinberg, M.S.; Margalit, R.; Leor, J. Targeting and modulating infarct macrophages with hemin formulated in designed lipid-based particles improves cardiac remodeling and function. J. Controll. Release 2017, 257, 21–31. [Google Scholar] [CrossRef]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, E.H.; Lee, J.H.; Park, E.H.; Park, H.E.; Jung, N.C.; Kim, T.H.; Koh, Y.S.; Kim, E.; Seung, K.B.; Park, C.; et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation 2017, 135, 1444–1457. [Google Scholar] [CrossRef]
- Tokutome, M.; Matoba, T.; Nakano, Y.; Okahara, A.; Fujiwara, M.; Koga, J.I.; Nakano, K.; Tsutsui, H.; Egashira, K. Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc. Res. 2019, 115, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.H.W.; Yang, K.Y.; Lui, K.O. An emerging role of regulatory T-cells in cardiovascular repair and regeneration. Theranostics 2020, 10, 8924–8938. [Google Scholar] [CrossRef]
- Li, J.; Yang, K.Y.; Tam, R.C.Y.; Chan, V.W.; Lan, H.Y.; Hori, S.; Zhou, B.; Lui, K.O. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics 2019, 9, 4324–4341. [Google Scholar] [CrossRef]
- Weaver, C.T.; Saparov, A.; Kraus, L.A.; Rogers, W.O.; Hockett, R.D.; Bucy, R.P. Heterogeneity in the clonal T cell response. Implications for models of T cell activation and cytokine phenotype development. Immunol. Res. 1998, 17, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, C.; Yang, K.Y.; Huang, X.; Han, M.Y.; Li, X.; Chan, V.W.; Chan, K.S.; Liu, D.; Huang, Z.P.; et al. Specific ablation of CD4(+) T-cells promotes heart regeneration in juvenile mice. Theranostics 2020, 10, 8018–8035. [Google Scholar] [CrossRef]
- Maldonado-Lasunción, I.; O’Neill, N.; Umland, O.; Verhaagen, J.; Oudega, M. Macrophage-Derived Inflammation Induces a Transcriptome Makeover in Mesenchymal Stromal Cells Enhancing Their Potential for Tissue Repair. Int. J. Mol. Sci. 2021, 22, 781. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, B.; Zhao, J.; Peng, Z.; Xu, W.; Yu, G. Effect of intravenous transplantation of hUCB-MSCs on M1/M2 subtype conversion in monocyte/macrophages of AMI mice. Biomed. Pharmacother. 2019, 111, 624–630. [Google Scholar] [CrossRef]
- Lee, T.M.; Harn, H.J.; Chiou, T.W.; Chuang, M.H.; Chen, C.H.; Chuang, C.H.; Lin, P.C.; Lin, S.Z. Preconditioned adipose-derived stem cells ameliorate cardiac fibrosis by regulating macrophage polarization in infarcted rat hearts through the PI3K/STAT3 pathway. Lab. Investig. 2019, 99, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhou, X.; Ge, Z.; Song, Y.; Wang, H.; Liu, X.; Zhang, D. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int. J. Biochem. Cell Biol. 2019, 114, 105564. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.I.; Kim, M.R.; Jeong, H.Y.; Jeong, H.C.; Jeong, M.H.; Yoon, S.H.; Kim, Y.S.; Ahn, Y. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp. Mol. Med. 2014, 46, e70. [Google Scholar] [CrossRef]
- Cho, D.I.; Kang, H.J.; Jeon, J.H.; Eom, G.H.; Cho, H.H.; Kim, M.R.; Cho, M.; Jeong, H.Y.; Cho, H.C.; Hong, M.H.; et al. Antiinflammatory activity of ANGPTL4 facilitates macrophage polarization to induce cardiac repair. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhao, N.; Zhang, J.; Liu, Y.; Zhu, D.; Kong, Y. Mesenchymal stem cells rejuvenate cardiac muscle through regulating macrophage polarization. Aging 2019, 11, 3900–3908. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Hu, J.; Chen, F.; Qiao, S.; Sun, X.; Gao, L.; Xie, J.; Xu, B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res. 2019, 115, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Zhang, F.; Chai, R.; Zhou, W.; Hu, M.; Liu, B.; Chen, X.; Liu, M.; Xu, Q.; Liu, N.; et al. Exosomes derived from pro-inflammatory bone marrow-derived mesenchymal stem cells reduce inflammation and myocardial injury via mediating macrophage polarization. J. Cell. Mol. Med. 2019, 23, 7617–7631. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Wu, J.; Cao, C.; Ma, L. Exosomes derived from regulatory T cells ameliorate acute myocardial infarction by promoting macrophage M2 polarization. IUBMB Life 2020, 72, 2409–2419. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Approach | Treatment | M2 Macrophage Phenotype | Reference |
|---|---|---|---|
| Cytokine treatment | IL-4 | CD206+F4/80+ | [77] |
| IL-10 | Arg1+Mrc1+Tgfb1+Ym1+ Fizz-1+ | [78] | |
| IL-6 | CD206+ | [79] | |
| Bioactive molecule treatment | Plasmin | Arg1+Ym1+Fizz-1+ | [80] |
| Angiotensin II | CD206+ | [82] | |
| Hydrogen sulfide | CD206+F4/80+ | [83] | |
| Hyaluronic acid-derived short oligosaccharides | CD206+ | [86] | |
| Collagen type I and type III matrices | CD206+MMP1+Arg1+ | [87] | |
| Plant-derived bioactive molecules | N-propargyl caffeate amide | M2a: CD163+FZZ1+YM-1+IL-10+Arg1+ | [88] |
| Resveratrol | CD206+F4/80+ | [90] | |
| Lipid particles with a drug | Hyaluronan lipid-based particles containing hemin | CD206+F4/80+ | [92] |
| Drug | SGLT2 Inhibitor Dapagliflozin | CD206+F4/80+CD68+IL-10+ | [93] |
| Regulation of the immune system | Activation of Treg by tolerogenic dendritic cells | CD68+MR+ | [96] |
| NF-κB inhibition by pioglitazone loaded into poly (lactic acid/glycolic acid) nanoparticles | Not specified | [97] | |
| Knock-out of CD226 | CD206+F4/80+ | [81] | |
| Stem cell therapy | Intravenous transplantation of hUCB-MSCs | CD11b+Ly6C- and F4/80+iNOS- | [103] |
| CD146+ MSCs | CD163+F4/80+ | [108] | |
| Exosomes | ADMSCs-derived exosomes | CD206+ | [105] |
| Mesenchymal stromal cell-derived exosomes | CD206+ iNOS- | [109] | |
| Exosomes derived from LPS-pre-conditioned BMSCs | CD206+ArgI+ | [110] | |
| Treg-derived exosomes | CD206+F4/80+Arg-1+TGF-β+ | [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. https://doi.org/10.3390/ijms22052715
Kim Y, Nurakhayev S, Nurkesh A, Zharkinbekov Z, Saparov A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. International Journal of Molecular Sciences. 2021; 22(5):2715. https://doi.org/10.3390/ijms22052715
Chicago/Turabian StyleKim, Yevgeniy, Sanzhar Nurakhayev, Ayan Nurkesh, Zharylkasyn Zharkinbekov, and Arman Saparov. 2021. "Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction" International Journal of Molecular Sciences 22, no. 5: 2715. https://doi.org/10.3390/ijms22052715