
Prolyl Hydroxylase 3 Knockdown Accelerates VHL-Mutant Kidney Cancer Growth In Vivo


Abstract
:1. Introduction
2. Results
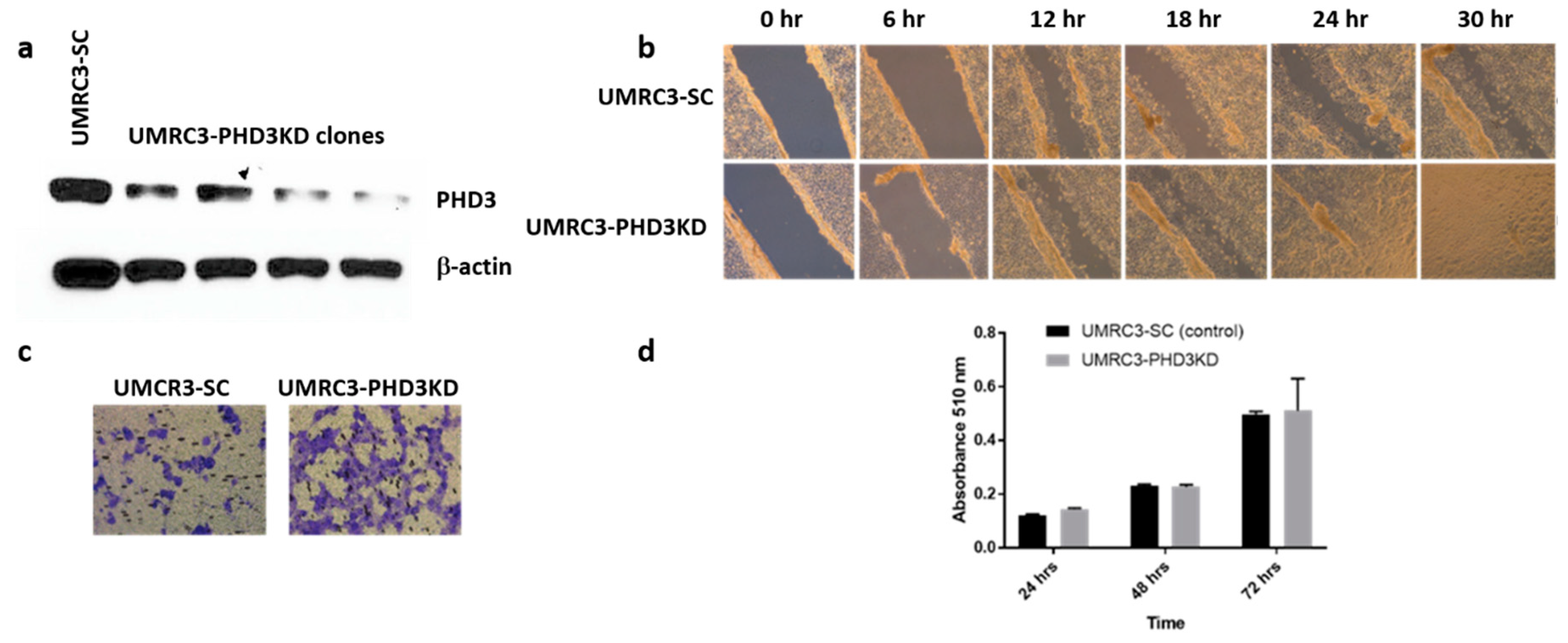
2.1. RCC Cell Lines Have Varying Levels of PHD3 Expression
2.2. PHD3 Knockdown Increases Cell Migration but Not Cell Proliferation
2.3. PHD3 Knockdown Increases Tumor Growth
2.4. PHD3 Knockdown Sensitizes Tumors to Sunitinib
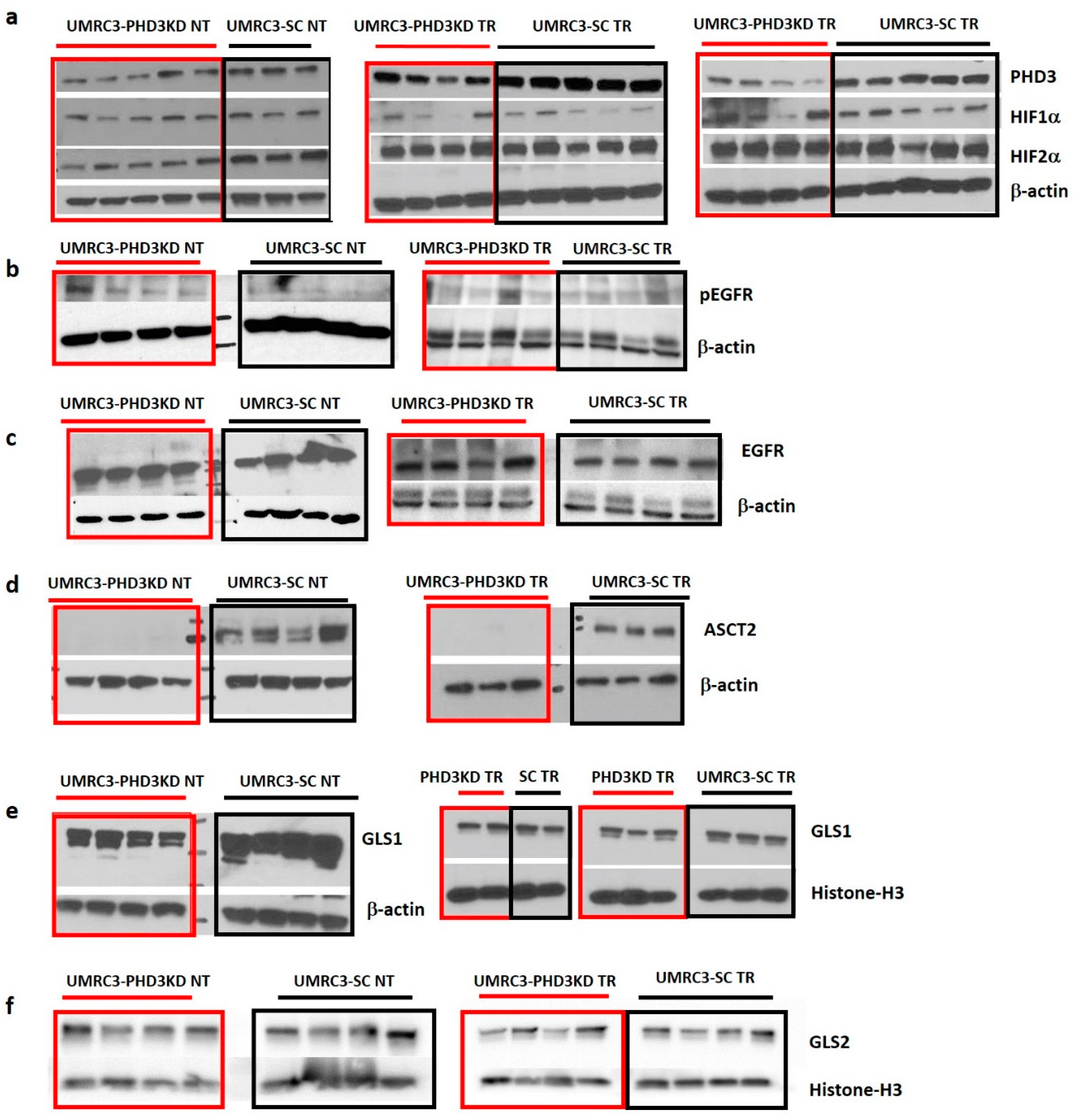
2.5. Varying HIF2α and pEGFR Expression with PHD3 Knockdown and Sunitinib Treatment
2.6. PHD3 Knockdown Changes Cellular Redox
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reverse Transcription Polymerase Chain Reaction
4.3. Lentiviral Transfection
4.4. Wound Healing Assay
4.5. Migration Assay
4.6. Cell Proliferation Assay
4.7. Hydrogen Peroxide Measurements in Cell Culture
4.8. Cell Proliferation Assay in the Presence of Hydrogen Peroxide
4.9. Mouse Models
4.10. GSH/GSSG Assay
4.11. Nuclear Magnetic Resonance (NMR) Based Metabolomics
4.12. Western Blotting and Densitometry
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ccRCC | clear cell renal cell carcinoma |
| HIF1α | hypoxia inducible factor 1 alpha |
| HIF2α | hypoxia inducible factor 2 alpha |
| PHD | prolyl hydroxylase |
| VHL | Von Hippel Lindau |
| NT | not treated |
| TR | treated with sunitinib |
| NMR | nuclear magnetic resonance |
| GLS1 | glutaminase 1 |
| ASCT2 | alanine-serine-cysteine transporter 2 |
| GSH | free glutathione |
| GSSG | oxidized GSH |
| EGFR | epidermal growth factor |
| pEGFR | phosphorylated EGFR |
| GLS2 | glutaminase 2 |
| H2O2 | hydrogen peroxide |
| ROS | reactive oxygen species |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | UMRC3-PHD3KD NT | UMRC3-SC NT | |||||
|---|---|---|---|---|---|---|---|
| Metabolites | Mean | SEM | N | Mean | SEM | N | Ratio |
| Valine | 0.048 | 0.009 | 5 | 0.050 | 0.015 | 4 | 1.0 |
| Lactate | 1.26 | 0.10 | 5 | 1.4 | 0.4 | 4 | 0.9 |
| Alanine | 0.176 | 0.019 | 5 | 0.15 | 0.04 | 4 | 1.2 |
| Glutamine | 0.026 | 0.004 | 5 | 0.012 | 0.005 | 4 | 2.2 |
| Acetate | 0.043 | 0.004 | 5 | 0.039 | 0.005 | 4 | 1.1 |
| Glutamate | 0.179 | 0.015 | 5 | 0.160 | 0.040 | 4 | 1.1 |
| Succinate | 0.029 | 0.002 | 5 | 0.025 | 0.007 | 4 | 1.2 |
| Fumarate | 0.0022 | 0.0002 | 5 | 0.0014 | 0.0003 | 4 | 1.6 |
| Aspartate | 0.016 | 0.002 | 5 | 0.009 | 0.003 | 4 | 1.7 |
| Triethylamine | 0.046 | 0.015 | 5 | 0.005 | 0.002 | 4 | 9.2 |
| Glutathione | 0.027 | 0.001 | 5 | 0.014 | 0.002 | 4 | 2.0 |



References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.S.; Goggins, W.B.; Yip, B.H.K.; Fung, F.D.H.; Leung, C.; Fang, Y.; Wong, S.Y.S.; Ng, C.F. Incidence and mortality of kidney cancer: Temporal patterns and global trends in 39 countries. Sci. Rep. 2017, 7, 15698. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricketts, C.J.; Maher, E.R. Renal Cell Carcinoma: Overview in Urological Oncology; Springer: London, UK, 2015; pp. 337–343. [Google Scholar]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Torigoe, T.; Hirohashi, Y.; Sato, E.; Honma, I.; Kitamura, H.; Masumori, N.; Tsukamoto, T.; Sato, N. Hypoxia-inducible factor (HIF)-independent expression mechanism and novel function of HIF prolyl hydroxylase-3 in renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2014, 140, 503–513. [Google Scholar] [CrossRef]
- Jaakkola, P.M.; Rantanen, K. The regulation, localization, and functions of oxygen-sensing prolyl hydroxylase PHD3. Biol. Chem. 2013, 394, 449–457. [Google Scholar] [CrossRef]
- Grossman, H.B.; Wedemeyer, G.; Ren, L.Q. Human renal carcinoma: Characterization of five new cell lines. J. Surg. Oncol. 1985, 28, 237–244. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Sunitinib: A novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag. 2007, 3, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Carroll, V.A.; Ashcroft, M. Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: Implications for targeting the HIF pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef] [Green Version]
- Garvalov, B.K.; Foss, F.; Henze, A.T.; Bethani, I.; Graf-Hochst, S.; Singh, D.; Filatova, A.; Dopeso, H.; Seidel, S.; Damm, M.; et al. PHD3 regulates EGFR internalization and signalling in tumours. Nat. Commun. 2014, 5, 5577. [Google Scholar] [CrossRef] [Green Version]
- Henze, A.T.; Garvalov, B.K.; Seidel, S.; Cuesta, A.M.; Ritter, M.; Filatova, A.; Foss, F.; Dopeso, H.; Essmann, C.L.; Maxwell, P.H.; et al. Loss of PHD3 allows tumours to overcome hypoxic growth inhibition and sustain proliferation through EGFR. Nat. Commun. 2014, 5, 5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miikkulainen, P.; Hogel, H.; Rantanen, K.; Suomi, T.; Kouvonen, P.; Elo, L.L.; Jaakkola, P.M. HIF prolyl hydroxylase PHD3 regulates translational machinery and glucose metabolism in clear cell renal cell carcinoma. Cancer Metab 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [PubMed]
- Wierzbicki, P.M.; Klacz, J.; Kotulak-Chrzaszcz, A.; Wronska, A.; Stanislawowski, M.; Rybarczyk, A.; Ludziejewska, A.; Kmiec, Z.; Matuszewski, M. Prognostic significance of VHL, HIF1α, HIF2α, VEGFA and p53 expression in patients with clear cell renal cell carcinoma treated with sunitinib as firstline treatment. Int. J. Oncol. 2019, 55, 371–390. [Google Scholar] [PubMed] [Green Version]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [Green Version]
- Miikkulainen, P.; Hogel, H.; Seyednasrollah, F.; Rantanen, K.; Elo, L.L.; Jaakkola, P.M. Hypoxia-inducible factor (HIF)-prolyl hydroxylase 3 (PHD3) maintains high HIF2α mRNA levels in clear cell renal cell carcinoma. J. Biol. Chem. 2019, 294, 3760–3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Donas, J.; Leandro-Garcia, L.J.; Gonzalez Del Alba, A.; Morente, M.; Alemany, I.; Esteban, E.; Arranz, J.A.; Climent, M.A.; Gallardo, E.; Castellano, D.E.; et al. Prospective study assessing hypoxia-related proteins as markers for the outcome of treatment with sunitinib in advanced clear-cell renal cell carcinoma. Ann. Oncol. 2013, 24, 2409–2414. [Google Scholar] [CrossRef]
- Beuselinck, B.; Verbiest, A.; Couchy, G.; Job, S.; de Reynies, A.; Meiller, C.; Albersen, M.; Verkarre, V.; Lerut, E.; Mejean, A.; et al. Pro-angiogenic gene expression is associated with better outcome on sunitinib in metastatic clear-cell renal cell carcinoma. Acta Oncol. 2018, 57, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Ma, Y.; Diaz de Leon, A.; Christie, A.; Xie, Z.; Woolford, L.; Singla, N.; Joyce, A.; Hill, H.; Madhuranthakam, A.J.; et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2019, 26, 793–803. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; Wehn, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ge, D.; Lu, C. The SMART App: An interactive web application for comprehensive DNA methylation analysis and visualization. Epigenetics Chromatin 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, X.; Liu, W.; Li, B.; Chen, D.; Hu, F.; Wang, L.; Liu, X.M.; Cui, R.; Liu, R. MicroRNA-1205, encoded on chromosome 8q24, targets EGLN3 to induce cell growth and contributes to risk of castration-resistant prostate cancer. Oncogene 2019, 38, 4820–4834. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Hoerner, C.R.; Chen, V.J.; Fan, A.C. The ‘Achilles Heel’ of Metabolism in Renal Cell Carcinoma: Glutaminase Inhibition as a Rational Treatment Strategy. Kidney Cancer 2019, 3, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hee, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, E.; Silva, C.; Martel, F. The role of the glutamine transporter ASCT2 in antineoplastic therapy. Cancer Chemother. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; van Geldermalsen, M.; Otte, N.J.; Lum, T.; Vellozzi, M.; Thoeng, A.; Pang, A.; Nagarajah, R.; Zhang, B.; Wang, Q.; et al. ASCT2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis 2017, 6, e367. [Google Scholar] [CrossRef]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutte, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Rahimi, F.; Broer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef] [Green Version]
- Broer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Broer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef] [Green Version]
- Bothwell, P.J.; Kron, C.D.; Wittke, E.F.; Czerniak, B.N.; Bode, B.P. Targeted Suppression and Knockout of ASCT2 or LAT1 in Epithelial and Mesenchymal Human Liver Cancer Cells Fail to Inhibit Growth. Int. J. Mol. Sci. 2018, 19, 2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, S.; von Eschenbach, A.C.; Giavazzi, R.; Fidler, I.J. Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 1986, 46, 4109–4115. [Google Scholar]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Voigt, W. Sulforhodamine B assay and chemosensitivity. Methods Mol. Med. 2005, 110, 39–48. [Google Scholar]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci. Rep. 2017, 7, 16159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]




| Tumor Type | PHD3/ β-actin | HIF1α/ β-actin | HIF2α/ β-actin | pEGFR/ β-actin | EGFR/ β-actin | ASCT2/ β-actin | GLS1/ β-actin | GLS1/ Histone-H3 | GLS2/ Histone-H3 |
|---|---|---|---|---|---|---|---|---|---|
| UMRC3-PHD3 NT | 0.38 ± 0.15 | 0.40 ± 0.11 | * 0.50 ± 0.15 | * 0.13 ± 0.04 | 5.58 ± 0.57 | * 0.0081 ± 0.00047 | 1.11 ± 0.26 | 1.04 ± 0.30 | |
| UMRC3-SC NT | 0.59 ± 0.03 | 0.44 ± 0.11 | * 0.95 ± 0.27 | * 0.05 ± 0.03 | 5.32 ± 0.87 | * 0.70 ± 0.32 | 1.50 ± 0.48 | 0.79 ± 0.32 | |
| UMRC3-PHD3 TR | * 0.21 ± 0.09 | 0.23 ± 0.19 | 0.92 ± 0.18 | 0.21 ± 0.15 | 0.50 ± 0.16 | * 0.019 ± 0.024 | 0.48 ± 0.13 | 0.83 ± 0.51 | |
| UMRC3-SC TR | * 0.54 ± 0.06 | 0.25 ± 0.10 | 0.77 ± 0.42 | 0.23 ± 0.07 | 0.52 ± 0.13 | * 0.81 ± 0.16 | 0.53 ± 0.07 | 0.77 ± 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacharias, N.M.; Wang, L.; Maity, T.; Li, L.; Millward, S.W.; Karam, J.A.; Wood, C.G.; Navai, N. Prolyl Hydroxylase 3 Knockdown Accelerates VHL-Mutant Kidney Cancer Growth In Vivo. Int. J. Mol. Sci. 2021, 22, 2849. https://doi.org/10.3390/ijms22062849
Zacharias NM, Wang L, Maity T, Li L, Millward SW, Karam JA, Wood CG, Navai N. Prolyl Hydroxylase 3 Knockdown Accelerates VHL-Mutant Kidney Cancer Growth In Vivo. International Journal of Molecular Sciences. 2021; 22(6):2849. https://doi.org/10.3390/ijms22062849
Chicago/Turabian StyleZacharias, Niki M., Lei Wang, Tapati Maity, Li Li, Steven W. Millward, Jose A. Karam, Christopher G. Wood, and Neema Navai. 2021. "Prolyl Hydroxylase 3 Knockdown Accelerates VHL-Mutant Kidney Cancer Growth In Vivo" International Journal of Molecular Sciences 22, no. 6: 2849. https://doi.org/10.3390/ijms22062849
APA StyleZacharias, N. M., Wang, L., Maity, T., Li, L., Millward, S. W., Karam, J. A., Wood, C. G., & Navai, N. (2021). Prolyl Hydroxylase 3 Knockdown Accelerates VHL-Mutant Kidney Cancer Growth In Vivo. International Journal of Molecular Sciences, 22(6), 2849. https://doi.org/10.3390/ijms22062849