Abstract
Iron is essential for multiple bacterial processes and is thus required for host colonization and infection. The antimicrobial activity of multiple iron chelators and gallium-based therapies against different bacterial species has been characterized in preclinical studies. In this review, we provide a synthesis of studies characterizing the antimicrobial activity of the major classes of iron chelators (hydroxamates, aminocarboxylates and hydroxypyridinones) and gallium compounds. Special emphasis is placed on recent in-vitro and in-vivo studies with the novel iron chelator DIBI. Limitations associated with iron chelation and gallium-based therapies are presented, with emphasis on limitations of preclinical models, lack of understanding regarding mechanisms of action, and potential host toxicity. Collectively, these studies demonstrate potential for iron chelators and gallium to be used as antimicrobial agents, particularly in combination with existing antibiotics. Additional studies are needed in order to characterize the activity of these compounds under physiologic conditions and address potential limitations associated with their clinical use as antimicrobial agents.
1. Introduction
Antimicrobial resistance is a significant burden on public health. The emergence of antibiotic resistance in multiple bacterial species affects many aspects of modern medicine, from the treatment of infections in the primary care setting to the clinical management of critically-ill patients receiving intensive care. The global dissemination of bacterial strains with resistance to multiple antibiotic classes represents a particular challenge to appropriate therapy, as in some cases there are few clinically-available antimicrobials that retain sufficient activity against these isolates [1]. Reports describing infections caused by pandrug resistant strains with resistance to all clinically-used antibiotics are especially worrisome since treatment options in these cases are severely limited [2]. The global health burden of antibiotic resistance is difficult to estimate. A report commissioned by the government of the United Kingdom in 2014 estimated that global deaths due to antimicrobial resistance could increase to 10 million per year, compared to an estimated 700,000 deaths in 2014, if current trends continue [3]. Although this report included multiple types of antimicrobial resistance and these long term projections are inherently difficult to quantify, the estimated impact of microbial resistance moving forward is clearly a cause for concern. This situation calls for the development of new antimicrobials with novel mechanisms of action. Unfortunately, very few antibiotics with completely novel mechanisms of action have been approved for clinical use over the last 40 years, and so there are currently very few new compounds in the development pipeline [4].
Given the continued emergence and dissemination of antimicrobial resistance, and the low likelihood that the need for new compounds with novel mechanisms of action will be met by the current development pipeline, exploring the potential of alternatives to traditional small molecule antibiotics is warranted. A recent report aiming to comprehensively evaluate alternatives to small molecule antimicrobials at the portfolio level identified metal chelation therapy (zinc, manganese and iron) as one of 19 approaches with potential for the treatment and prevention of antibiotic resistant infections [5]. Over the last decade there has been special interest in targeting bacterial iron metabolism as an antimicrobial strategy given the necessity of iron for the growth and survival of most pathogenic bacterial species. Iron chelation and gallium-based therapies have been broadly studied with multiple different bacterial species, most notably with pathogenic species that are associated with multidrug resistant infections. Studies ranging from the in-vitro characterization of the antimicrobial activity of different chelators to in-vivo studies in animal models of infection have shed light on the potential of iron chelators as potential antimicrobial agents. In this review, we provide a synthesis of published studies that have evaluated the antimicrobial activity of iron chelating molecules and gallium-based therapies on bacterial species associated with antibiotic resistance, with particular emphasis on recent studies with novel compounds, and studies that have evaluated potential synergies with existing antimicrobials. In addition, we provide an overview of the challenges that remain to be addressed before iron chelation therapy could be used clinically. Approaches other than iron chelation and gallium-based therapies that target bacterial iron metabolism, such as inhibiting siderophore biosynthesis and assimilation, antibiotic-siderophore conjugates, and inhibiting heme assimilation have been explored and are reviewed elsewhere [6,7].
2. Iron Acquisition in Pathogenic Bacteria
Iron is required by most bacterial pathogens for growth and survival, and is therefore essential during the establishment of host colonization and infection. Many species have evolved to use iron in multiple physiological processes including, DNA synthesis, transcription and cellular respiration, likely due to its high abundance in many natural environments (it is one of the most abundant elements on earth) and its readily exploitable oxidation/reduction chemistry. However, in contrast to natural environments and bacterial growth media, free iron in the human body is maintained at a very low concentration, approximately 10−24 M [8], due to potential damage to the host caused by the high reactivity of non-complexed iron. Maintaining free iron at very low concentrations also serves to strengthen host defense against infection by contributing to what has been termed nutritional immunity [9]. Low free iron concentrations in the body are achieved by the activity host proteins that form strong complexes with iron. Approximately 70% of total body iron is found in red blood cells in the form of hemoglobin-bound heme complexes [9]. Ferric (Fe(III)) iron is also sequestered by the host proteins transferrin and lactoferrin, which exhibit very high iron affinities with binding constants of ~10−20 M and ~10−23 M, respectively [10,11]. Lactoferrin is secreted during the innate immune response and maintains its high affinity or iron in the acidic environment created by infection, thus promoting nutritional immunity [12].
Pathogenic bacteria have evolved mechanisms to obtain host iron that can be broadly classified into two groups; (i) siderophore-mediated iron acquisition, and (ii) specific acquisition mechanisms that obtain iron from complexed host proteins such as heme, transferrin and lactoferrin. These mechanism are briefly summarized here; however, the reader is referred to a number of excellent monographic reviews covering these processes [9,13,14,15]. As a general mechanism, bacterial siderophores are extracellular molecules with very high affinity for ferric iron (with binding constants of 1050 M−1 in some cases) that are secreted by many different pathogenic Gram positive and Gram negative species (Figure 1). The high affinity of siderophores allows them to strip iron from host proteins, after which the siderophore-iron complex is internalized via a siderophore-specific receptor on the bacterial cell surface [6]. Upon reaching the bacterial cytosol, ferric iron is released to the intracellular iron pool either through cleavage of the siderophore or through the reduction of Fe(III) to Fe(II), which lowers binding affinity to the siderophore thus permitting its dissociation [9]. It is important to note that this general mechanism does not apply to all siderophore systems, as it has been shown that the Pseudomonas aeruginosa siderophore pyoverdine releases iron in the periplasm before transport to the cytosol [16].
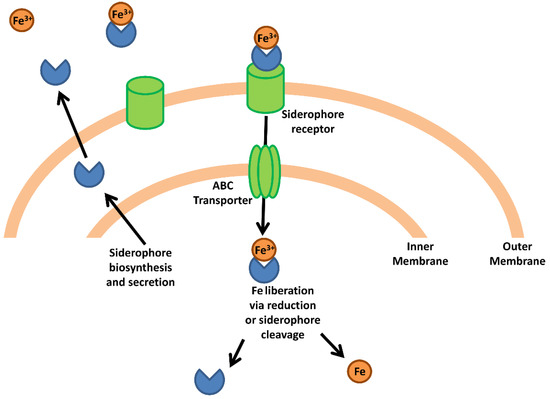
Figure 1.
Bacterial iron acquisition via siderophores. The schematic represents a generic siderophore-mediated iron acquisition system based on common features in Gram-negative species. Siderophores are synthesized and secreted from the bacterial cell where they capture free iron or strip iron complexed to host proteins. The siderophore-iron complex binds a siderophore-specific receptor on the bacterial cell surface and is transported through the extracellular membrane. An ABC transporter transports the siderophore-iron complex into the cytoplasm where the complex is dissociated via reduction or enzymatic cleavage of the siderophore, releasing the iron atom into the intracellular iron pool.
Acquiring iron from host transferrin and lactoferrin occurs via the interaction between these proteins, complexed with ferric iron, and specific receptors on the surface of bacterial cells. These receptors are able to strip Fe(III) from transferrin and lactoferrin and transport it to the interior of the cell. In the case of heme iron, multiple secreted bacterial effectors have been shown to lyse erythrocytes in order to liberate heme and its associated ferrous (Fe(II)) iron. In addition, many bacterial pathogens secrete hemophores, which capture heme from host hemoglobin and shuttles it to receptors on the bacterial outer membrane [17]. These specific receptors on the bacterial cell surface bind these heme complexes which are then transported to the cytosol. Bacterial enzymes, such as heme monooxygenase, cleave the heme moiety and release the Fe(II) into the intracellular iron pool. The acquisition of ferrous iron can also be facilitated by specific uptake systems, such as the Feo system, which is broadly distributed in different Gram negative species [18].
3. Iron Chelators as Antimicrobial Agents
Given the absolute necessity of iron for the growth and survival of many pathogenic microorganisms, decreasing available iron at the site of infection has potential to contribute to treatment approaches. One strategy for achieving iron limitation is the use of chelating molecules that sequester the metal and prevent its uptake by the microorganism causing infection. Iron chelators can bind iron in both its ferrous and ferric states; however, they typically have greater affinity for one or the other [19,20]. In addition, some chelators may also be able to bind other metal ions such as copper(II), zinc(II) or gallium(III). The ability of these compounds to chelate Fe(III) with greater stability and affinity depends on the ligands available for iron coordination. Within chelator-iron complexes, Fe(III) is coordinated by forming an octahedral structure next to six donor atoms in which the metal remains in the center. These ligands can be classified according to whether they have two (bidentate), three (tridentate) or six (hexadentate) donor atoms available for coordination with iron [20]. The stability of the chelator-iron complex improves with more donor atoms available in the ligand, with the hexadentates demonstrating the greatest stability. This may explain why most siderophores produced by microorganisms use hexadentate coordination.
The antimicrobial activity of iron chelators has been studied and demonstrated for decades [21,22]. In the following sections, we summarize published studies evaluating the activity of different iron chelators against the most problematic antibiotic resistant bacterial species. Chelators have been grouped according the chemical moiety that participates in coordination of Fe(III) and include; hydroxamates, aminocarboxylates, hydroxypyridinones and cathecols (Figure 2).
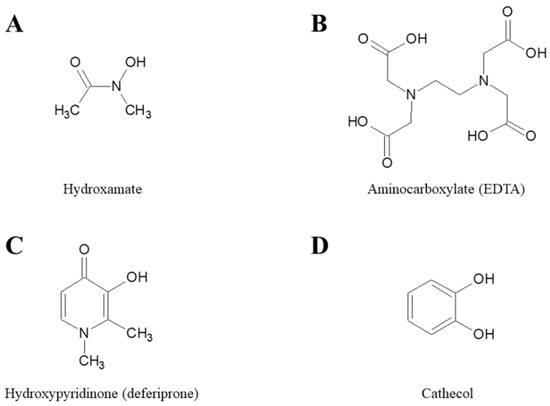
Figure 2.
Chemical structures of the parent molecules of different classes of iron chelators. (A) Hydroxamate, (B) Aminocarboxylate, (C) Hydroxpyridinone, and (D) Cathecol.
3.1. Hydroxamates
Compounds derived from hydroxamic acid (Figure 2A) have high affinity for Fe(III). Many siderophores produced by microorganisms, including the prototypical enterobactin from Escherichia coli, contain hydroxamate moieties [8]. One of the first iron chelators to be evaluated as an antimicrobial was deferoxamine (DFO), a hexadentate trihydroxamate siderophore produced by Streptomyces pilosus that is FDA-approved for the treatment of thalassemia and other iron disorders. DFO can inhibit the growth of Staphylococcus aureus, Pseudomonas aeruginosa and Acinetobacter baumannii at concentrations between 2.5 and 10 mg/mL in Mueller-Hinton broth, a medium with high iron content (Table 1) [23]. In another study, DFO was tested against these same pathogens, in addition to Escherichia coli and Kleblsiella pneumonia, with no inhibition observed at concentrations ≤512 mg/L in Mueller-Hinton broth and in iron-poor RPMI 1640 [24]. DFO has also demonstrated efficacy in reducing the formation of biofilm in a cellular model of cystic fibrosis by 42% in P. aeruginosa at 400 mg/L (approximate 0.71 mM) [25]. However, in the same work, DFO failed to inhibit established biofilms in these cells and in an abiotic plastic surface at the same concentration [25]. This is in line with a study by Banin et al. [26], which describes no effect of DFO either against biofilm formation (at 0.001 mM) and established biofilms (at 1 mM).

Table 1.
In vitro antimicrobial activity of iron chelators.
Another natural compound, the siderophore produced by Mycobacterium smegmatis exochelin-MS (Exo-MS), has been tested for antimicrobial activity against S. aureus, P. aeruginosa and A. baumannii and was more inhibitory at lower concentrations than DFO (minimum inhibitory concentrations (MICs) between 0.05 and 0.125 mg/mL) [23].
A potential limitation of microorganism-derived compounds is the possibility that other microbial species can recognize these xenosiderophores, internalize them and exploit the associated iron. Different pathogens, such as P. aeruginosa, S. aureus or E. coli, have been shown to be able to use iron-DFO complexes as an iron source [37,38,39,40]. In fact, Visca et al. described the capability of siderophore null mutant P. aeruginosa to grow in an iron-poor media at suboptimal concentration of DFO (20 µM) better than without it [41]. Also, in a study by de Leseleuc et al. [42], the growth of A. baumannii in RPMI supplemented with 10% fetal bovine serum was encouraged by the presence of DFO. This may explain the high MICs seen with DFO against these microorganisms. However, the ability of DFO to be taken up by bacteria that do not produce it has been revealed as an effective strategy to transport different molecules with antimicrobial activity into these pathogens [26,43].
3.2. Aminocarboxylates
Aminocarboxylates (Figure 2B) include a wide variety of compounds known for their chelating capacity, such as ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA). They have high affinity for Fe(III) but generally low selectivity, which can cause toxicity, such as zinc depletion described in patients treated with DTPA [44].
EDTA activity has been evaluated most notably against P. aeruginosa and has been shown to decrease the formation of biofilms by this microorganism. In a study by Banin et al. [45], a concentration of 50 mM was able to reduce the number of cells associated with biofilms by >99%. Furthermore, this activity is inversely proportional to the concentration of iron or calcium in the medium, demonstrating that metal chelation is involved in its mechanism of action. In a separate study, higher concentrations of EDTA (30 mg/mL; approximately 100 mM) were required to reduce the cell count associated with biofilms by approximately 2 log10 [35]. Different laboratory conditions, as well as the P. aeruginosa strain used, may contribute to the difference observed. EDTA can significantly inhibit the growth of P. aeruginosa in both aerobic and anaerobic conditions at concentrations of 1250 µM and 650 µM, respectively, and impair the biofilm formation to 312 µM [36]. EDTA has also been combined with phenyl-arginine-β-naphthylamide (PAβN), an inhibitor of the MexAB-OprM efflux system in P. aeruginosa which is overexpressed when the bacterium grows under severe iron limiting conditions [46]. The combination of EDTA with 50 µg/mL PaβN is able to significantly inhibit planktonic growth at low concentrations (2.5 µg/mL), and even block it completely at 160 µg/mL [47]. The combination of 5 µg/mL EDTA with PaβN also inhibits the formation of biofilm in P. aeruginosa [47].
DTPA has also shown antimicrobial activity against different species in vitro. O’May et al. describe activity similar to EDTA in inhibiting the growth and biofilm formation of P. aeruginosa in both aerobic and anaerobic conditions at concentrations of 1250 µM and 650 µM, respectively [36]. DTPA can also inhibit the growth of S. aureus (bacterial reduction of 97.3% at 500 µg/mL), P. aeruginosa (bacterial reduction of 86.2% at 100 µg/mL) and E. coli (bacterial reduction of 98.5% at 250 µg/mL) [30].
3.3. Hydroxypyridinones
Hydroxypyridinones (HPO) (Figure 2C) are a group of synthetic compounds that have affinity for Fe(III) similar to that of cathecols, and the stability of the iron-chelator complexes are similar to that described for hydroxamates. However, unlike these two types of ligands, the chemical structure of HPOs does not resemble ligands that are found in the bacterial siderophores. This difference in structure makes Fe(III)-HPO complexes difficult to capture and exploit by pathogenic microorganisms, in contrast to, for example, DFO. Within the different types of HPO, the 3-hydroxypyridin-4-ones are the ligands of this group that have the highest affinity for Fe(III) [19].
Deferiprone (1,2-dimethyl-3-hydroxypyridin-4-one; DFP) is a bidentate ligand with high affinity for Fe(III) and has been approved for the treatment of iron overload in patients with thalassemia [48]. This compound has moderate activity in vitro against Gram positive and Gram negative species. The growth of S. aureus and other coagulase-negative staphylococci is inhibited in the presence of 1.5 mM DFP (approximately 200 µg/mL) after 6 h of incubation, although S. aureus regrowth at the same level as the control culture occurred after 24 h [49]. In a separate study by Thompson et al. the in vitro activity of DFP is evaluated against important nosocomial pathogens, demonstrating minimal inhibitory concentrations (MICs) of 128–512 µg/mL for A. baumannii, E. coli, Klebsiella pneumoniae and P. aeruginosa in cation-adjusted Mueller-Hinton broth. When the same assay is performed in the iron-poor culture medium RPMI 1640, the MICs are 2–4 fold lower in some cases. DFP is not able to inhibit the growth of S. aureus [24]. Consistent with these results, de Léséleuc et al. obtain similar results with A. baumannii in the presence of DFP with a MIC of 128 µM in media with high and low iron content [42]. Although the antimicrobial activity of DFP seems to be mainly related to its iron chelating capability, Visca et al. suggest that there must be a chelation-independent toxicity since the addition of iron reverse but does not prevent the antimicrobial activity at high concentrations of DFP (≥3.67 mM) [41].
As previously described for deferoxamine, DFP can act as a growth promoter for some bacterial species, like P. aeruginosa and A. baumannii, at sub-inhibitory concentrations in iron-limiting conditions [41,42]. Moreover, DFP can capture Fe(III) bound to proteins such as transferrin [50], and is able to mobilize it from these iron sources thus facilitating its uptake by A. baumannii [42].
Another HPO with antimicrobial activity is cyclopirox (6-cyclohexyl-1-hydroxy-4-methylpyridin-2-one), a topical antifungal used for decades in the treatment of superficial mycoses. Although its mechanism of action is not fully characterized, studies carried out with Candida albicans have revealed changes in the expression of genes similar to those that occur in conditions of iron limitation [51,52]. Furthermore, its activity decreases with the addition of iron to the culture [51,52]. In a study by Carlson-Banning et al., cyclopirox is able to inhibit the growth of E. coli (MICs 5–15µg/mL), K. pneumoniae (MICs 5–15µg/mL), A. baumannii (MICs 5–7 µg/mL), and P. aeruginosa (MICs 10 - >30 µg /mL) with differing antibiotic resistance profiles [27].
As mentioned above, hexadentate ligands have higher affinity for Fe(III) than bidentate ligands with similar chemical structures. For this reason, hexadentate compounds derived from HPO have been designed with the aim of achieving Fe(III) binding constants similar to that of bacterial siderophores in order to compete with them for iron sequestration. The hexadentate compound CP251 is able to completely inhibit the in vitro growth of S. aureus and P. aeruginosa at 500 µg/mL and 100 µg/mL, respectively [30]. It also decreases the growth of E. coli 2log10 at 250 µg/mL (bactericidal rate of 99.6%) [30]. Several studies have analyzed various hexadentate HPOs with results similar to Qiu et al. [53,54].
3.4. DIBI
The development of hexadentate HPOs has also focused on the design of larger compounds with less systemic absorption than smaller molecules in order to avoid some of the adverse effects derived from iron depletion in certain biological compartments. Polymers or dendrimers (branched polymers) derived from HPO may have significant antimicrobial activity and be effective in treating localized infections, such as wound infections or infections of the respiratory mucosa. One example of this type of compound is DIBI [55], a 9 kDa copolymer that has nine broadly spaced hydroxypyridinone metal-binding groups that can coordinate with three iron molecules with full hexadentate binding (Figure 3). DIBI is highly water soluble, shows selective binding to Fe(III) ions and has not demonstrated toxicity in preclinical animal models [55]. In recent years, multiple studies have demonstrated the antimicrobial activity of DIBI against multiple antibiotic resistant bacterial species including both Gram positive and Gram negative pathogens. Initial studies with DIBI demonstrated strong antimicrobial activity against antibiotic susceptible reference strains for S. aureus (MIC; 4 µg/mL), A. baumannii (MIC; 2 µg/mL) and C. albicans (MIC; 2 µg/mL). These MIC values were one to two log10 lower than MIC values observed for previously the characterized iron chelators DFP and DFO, respectively. Importantly, given the large size of DIBI compared to these chelators, the molecular weight of DIBI is 9 kDa vs. 139 Da for DFP, these MICs translate into very low concentrations of the polymer.
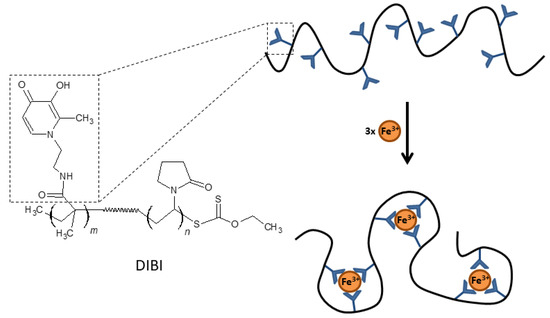
Figure 3.
Chemical structure and schematic representation of Fe(III) binding by DIBI.
In a separate study, DIBI also demonstrated activity against methicillin resistant S. aureus isolates, with MICs between 1 and 4 µg/mL [32]. DIBI also showed activity in vivo against S. aureus in a wound infection model, resulting in reduced bacterial loads and tissue inflammation compared to control mice, and intranasal administration of DIBI was also able to reduce nasal S. aureus carriage by approximately two log10 compared to control mice [32]. In a separate study, DIBI was able to reduce nasal S. aureus carriage of both mupirocin susceptible and resistance isolates in a mouse model [33]. Importantly, no post-treatment resistance to DIBI was observed. DIBI demonstrates strong antimicrobial activity in vitro against clinical isolates of A. baumannii, with MIC values of 4 µg/mL [28]. Intranasal administration of DIBI was also able to reduce tissue bacterial loads, reduce dissemination to the spleen and improve survival in a mouse pulmonary infection model [28].
In addition to its antimicrobial activity, data from preclinical experimental models have indicated that DIBI has anti-inflammatory activity during sepsis [56,57]. Treatment with DIBI was able to reduce inflammation-associated markers (leukocyte activation, loss of capillary perfusion and tissue damage) after administration of lipotechoic acid from S. aureus and lipopolysaccharide from E. coli and K. pneumoniae [56]. Although the molecular mechanisms underlying the anti-inflammatory activity observed with DIBI are not fully understood, it has been hypothesized that is may be due to reduced iron-catalyzed reactive oxygen species production by leukocytes during sepsis [56,57]. Taken together, these studies indicate that DIBI may be a promising antimicrobial compound.
3.5. Other Iron Chelators
There are additional chelating compounds that can bind iron with high affinity. Some of the first molecules tested against microorganisms have a natural origin, as is the case for the human proteins lactoferrin and transferrin. Both compounds can inhibit the formation of biofilms in P. aeruginosa [58,59,60]. Their in vitro activity against S. aureus, A. baumannii, K. pneumoniae and C. albicans has also been described [61,62]. However, some pathogens, such as P. aeruginosa, are able to use these proteins as an iron source [63], which limits their efficacy as antimicrobials against these microorganisms. Compounds derived from cathecol have high affinity for Fe (III) due to the high electron density generated by the two oxygen atoms (Figure 2D) [20]. Cathecol moieties are a fairly common motif in bacterial siderophores. However, this electron charge density also gives them high affinity for protons, making these compounds less stable within the physiological pH range [20]. Compounds derived from 2,3-dihydroxybenzoic acid have been show to inhibit the formation of P. aeruginosa biofilms [64,65].
Deferasirox (DFX) is a triazole-derived trivalent chelator that was approved by the FDA in 2005 for the treatment of iron overload. DFX can inhibit antibiotic resistant strains of S. aureus with a MIC and a minimum bactericidal concentration of 50 mg/L [34]. It is also able to reduce in vitro biofilm formation of P. aeruginosa by 99% at a concentration of 1 µM after 4 h of treatment [25]. However, the use of DFX is limited by its toxicity [66].
The chelator 2,2′-bypiridyl (BIP), which can cross biological membranes and exerts its action in the cytoplasm of cells, has been widely studied in vitro. It should be noted that it has greater affinity for Fe(II) than for Fe(III). BIP inhibits the growth of pathogens such as K. pneumoniae, E. coli, A. baumannii, S. aureus and P. aeruginosa with MICs between 64 and 512 µg/mL [24,29], and can inhibit the formation of P. aeruginosa biofilm [36]. The therapeutic use of BIP is limited by its neurotoxicity [67]. However, this chelator is used in the laboratory to simulate iron-limiting conditions experimentally [41,68,69].
Finally, derivatives of 8-hydroxyquinoline have also been explored as antimicrobial iron chelators. An example of this type of compound is nitroxoline (5-nitro-8-hydroxyquinoline), a well-known antimicrobial used for the treatment of urinary tract infections. It can chelate both divalent and trivalent metals and this chelation is pH dependent [70]. Nitroxoline inhibits the planktonic growth of P. aeruginosa, K. pneumoniae and E. coli [31]. It can also reduce biofilm formation by up to 80% and decrease 4log10 the number of viable cells in preformed P. aeruginosa biofilms at concentrations of 8 and 200 µg/mL, respectively [31]. Another 8-hydroxyquinoline derivative is VK28, which demonstrates in-vitro activity against important nosocomial pathogens when the growth medium is poor in iron [24].
4. Gallium
The therapeutic properties of gallium, specifically Ga(III), are due to its physical-chemical similarity to Fe(III) (reviewed in [71,72]). Unlike Fe(III), Ga(III) cannot be reduced under physiological conditions. This inability to participate in redox reactions likely inhibits the catalytic activity of numerous enzymes and blocks essential functions of the bacterium. However, the specific molecular mechanisms of action responsible for the antimicrobial activity of Ga(III) have not been fully characterized. Recently, Wang et al. demonstrated Ga(III) binding to two subunits of RNA polymerase in P. aeruginosa, potentially explaining RNA synthesis inhibiting activities [73]. Due to its ability to alter iron metabolism, the antimicrobial potential of various compounds derived from gallium has been studied in multiple studies in recent years.
One of the first compounds tested was gallium nitrate (Ga(NO3)3), which was approved by the FDA for treatment of cancer-associated hypercalcemia. The activity of Ga(NO3)3 is moderate against A. baumannii regardless of the iron concentration of the medium [42,68]. However, the activity improves when serum is added to the medium or if it is cultivated directly on serum-containing solid media [42,68,74]. This effect is dose and strain dependent, and 90% growth inhibition is achieved at concentrations between 3 and 64 µM (Table 2). Ga(NO3)3 also inhibits the formation of A. baumannii biofilm in human serum at 16 µM, and can disrupt preformed biofilms at 64µM [74]. This increased activity could be due to a synergistic effect with transferrin, which may chelate Ga(III) and act as a platform for gaining access to the interior of bacterial cells. Furthermore, there is evidence that the presence of this protein can induce expression the iron uptake system in A. baumannii [75]. An increase in production of siderophores may facilitate the uptake of Ga(III), as seen in other species [76,77]. The use of Ga(NO3)3 has also been evaluated against S aureus, and is able to significantly inhibit the growth of planktonic cells and preformed biofilms at ≥16 µM and ≥128 µM, respectively [78].
Table 2.
In-vitro antimicrobial activity of Gallium compounds.
Gallium-protoporphyrin IX (GaPPIX) is another compound that has been evaluated as an antimicrobial. This non-iron heme derivative uses heme-uptake pathways to reach the interior of the cell and inhibit hemoproteins such as cytochromes, catalases or peroxidases [85]. Hizaji et al. have described that GaPPIX inhibits the growth of P. aeruginosa (IC50 = 12 µM) by targeting heme-dependent b-type cytochromes [84]. GaPPIX also inhibits different strains of A. baumannii at concentrations ≤20 µM [81]. Based on the idea that the access route that GaPPIX exploits is different from that of Ga(NO3)3, Choi et al. evaluated the combined use of these compounds against different nosocomial pathogens [79]. The combination of GaPPIX and Ga(NO3)3 demonstrated a synergistic effect against P. aeruginosa, A. baumannii, K. pneumoniae and S. aureus with fractional inhibitory concentrations index (FICI) of 0.5, 0.5, 0.13 and 0.37, respectively; with an FICI of ≤0.5 considered as a synergistic effect between two compounds against a specific microorganism. Furthermore, this synergy was also reflected in the disruption of K. pneumoniae and P. aeruginosa biofilms (>90% and >95%, respectively, compared to the positive control) at concentrations in which they individually have no activity [79].
Other compounds derived from gallium such as gallium maltolate (GaM) and gallium citrate (GaCi) have been tested against bacteria such as S. aureus or K. pneumoniae [82,83]. In a study by Hijazi et al. the in-vitro activity of GaM, GaPPXI and Ga(NO3)3 against different antibiotic resistant nosocomial pathogens in different Fe(III) concentration and in a medium supplemented with human serum (RPMI-HS) were determined [80]. The susceptibility of these microorganisms to different gallium compounds varied between species and strains. Susceptibility was greater in iron-poor conditions (like RPMI medium supplemented with serum) with GaM and Ga(NO3)3, and GaPPIX was the only compound with activity in serum-free media, regardless of Fe(III) concentration. This may be explained by the different systems employed for heme-uptake in these species, and by the presence of albumin in serum which may block the action of GaPPIX [80].
Finally, gallium-derived compounds have also shown efficacy in vivo. Both Ga(NO3)3 and GaPPIX increased the survival of Galleria mellonella in models of A. baumannii infection [68,81]. Ga(NO3)3 is also active in models of P. aeruginosa and A. baumannii lung infection in mice [42,86]. Moreover, there is a phase 1b human trial whose results have been published recently [87]. In this study, the antibiotic activity of gallium in patients with cystic fibrosis and chronic P. aeruginosa airway infections was evaluated. Ga(NO3)3 was administered by slow intravenous infusion over 5 days in two different dose regimens (100 and 200 mg/m2/day). No serious adverse effects were observed, and plasma and sputum gallium levels remained detectable for prolonged periods. Remarkably, these patients presented a statistically significant increase in lung function in a magnitude similar to that produced by approved antimicrobials in cystic fibrosis. In addition, Ga(NO3)3 treatment decreased sputum P. aeruginosa concentrations, but they were not statistically significant. There are two other human trials in which gallium antimicrobial activity has been evaluated in cystic fibrosis patients: a phase 2 study for patients with cystic fibrosis infected with P. aeruginosa (IGNITE study) and a phase 1 study for patients with cystic fibrosis who are colonized with nontuberculous mycobacterias (ABATE study) (www.clinicaltrials.gov accessed on 9 February 2021).
5. Iron Chelators and Gallium Combinations
As previously seen, some iron chelators might act as carriers for other antimicrobial compounds within bacteria. In this way, Ga(III) has also been tested in combination with deferoxamine. DFO is captured by bacterial species such as S. aureus or P. aeruginosa, potentially acting as a Ga(III) carrier. DFO-Ga(III) can inhibit the planktonic growth of P. aeruginosa [26] and E. coli [43] better than the Ga(III) alone; while it does not appear to have activity against S. aureus [43]. Moreover, DFO-Ga(III) blocks P. aeruginosa biofilm formation at planktonic subinhibitory concentrations (0.001 mM) and cause a 3–4 log10 decrease in cell count of P. aeruginosa established biofilms [26].
Another combined strategy has been the use of DFP together with GaPPIX. These two compounds applied sequentially (first DFP and then GaPPIX) significantly reduced preformed S. aureus biofilms compared to the compounds used separately (94% vs 77% of de most active single compound) [88]. However, in another study by Richter et al., the formulation of DFP and GaPPXI in gel form failed to have a greater effect against S. aureus biofilm compared to GaPPXI alone [89]. In this case, there was a significant overall effect against a P. aeruginosa biofilm (2 log10 reduction compared to GaPPXI alone) [89].
6. Iron Chelators and Gallium in Combination with Antimicrobials
As described previously, alteration of iron homeostasis has a deleterious effect on bacterial growth. In addition to this inhibition per se, a decrease in intracellular iron concentrations can enhance the activity of other antimicrobials. Thus, combined treatment with currently-used antibiotics and iron chelators or Ga(III) compounds could have synergistic effects.
Some iron chelators such as DFO or DFX have been evaluated together with other antimicrobials [25,34]. DFX can increase vancomycin binding to the cell surface and together they have greater activity in vitro and in vivo against S. aureus than each compound used separately [34]. In a study by Moreau-Marquis et al. the combined use of DFO or DFX with tobramycin decreased the biomass of preformed P. aeruginosa biofilms by 90% [25]. DFX and tobramycin used together also significantly decrease the formation of P. aeruginosa biofilms [25].
Compounds derived from HPO also increase the activity of other antimicrobials. One example is DIBI, the copolymer derived from 3-hydroxypyridin-4-one [55]. This compound has shown in-vitro synergy with multiple antimicrobials such as gentamicin and ciprofloxacin against A. baumannii and S. aureus, including isolates resistant to the co-administered antibiotic [28,32,33].
There are also several studies testing Ga(III) derived compounds together with other antimicrobials. Antunes et al. demonstrated that Ga(NO3)3 acts synergistically with colistin against A. baumannii (FICI values ranging from 0.13 to 0.5) [68]. The alteration of the outer membrane produced by colistin probably favors the diffusion of Ga(III) into the cell [68]. Another study also describes synergy between Ga(NO3)3 and GaPPIX together with colistin, rifampin or ciprofloxacin against K. pneumoniae, P. aeruginosa and S. aureus [79]. Finally, the gel formulation of DFP and GaPPXI effective against P. aeruginosa biofilms when used alone demonstrated improved activity when used together with ciprofloxacin [89].
7. Challenges to Developing Iron Chelation/Gallium-Based Therapies
Although iron chelation and gallium-based therapies have demonstrated antimicrobial activity in-vitro and in a handful of in-vivo studies, multiple challenges are associated with the development of these therapies for clinical use. First, during preclinical development of these compounds, the antimicrobial activity observed with iron chelators and gallium compounds is highly dependent upon the conditions used for their characterization. Given that these compounds target bacterial iron metabolism, the concentration of free iron in the assays used for quantifying activity can significantly affect results. As can be appreciated in Table 1, multiple assay conditions have been employed, most typically bacterial growth medium (such as Mueller-Hinton broth) and cell culture media (RPMI). Some of these assay conditions may not reflect physiologically relevant iron concentrations, thus altering the observed antimicrobial activities. This may be especially true for iron rich laboratory growth media. As described in the preceding sections, growth media can significantly influence the observed antimicrobial activity of iron chelating and gallium compounds. In addition to iron, concentrations of other polyvalent cations such as zinc and manganese, can potentially affect the activity since it is known that some chelators have high affinity for these metals. For these reasons, the use of media that accurately reflect physiological ion concentrations may be desirable in initial in-vitro assays assessing chelator or gallium activity. An additional important consideration is the strain-dependent variation of the activity of iron chelators and gallium-based compounds. Initial in-vitro studies characterizing the activity these compounds often employ antibiotic susceptible reference strains, which may not be representative of antibiotic resistant clinical isolates that produce disease. Many of the studies included in this review have characterized activity against antibiotic resistant isolates [23,24,28,32,33,68,80], a critical aspect required for appropriately evaluating the therapeutic potential of these compounds. In-vivo studies can provide highly relevant information on chelator/gallium activity as these models are physiological with respect metal ion concentrations, and also account for aspects such as compound biodistribution, pH and the presence of host factors.
A second aspect that must be addressed for iron chelators and gallium-based compounds is elucidating the mechanisms by which they affect existing antimicrobials. One of the most likely uses of these compounds is in combination therapy together with existing antibiotics; however, very little is known regarding how chelators and gallium interact with currently approved antimicrobials. The activity seen with some combinations of chelators/gallium and antibiotics (described above) may reflect the summative effects of the antibiotic with reduced bacterial growth/survival in the absence of available iron. However, there is evidence indicating that the additive effects of these chelator/antibiotic combinations may be more complex. Free iron concentrations can alter bacterial transcriptional programs, most notably through the Fur protein [90]. Under low iron concentrations, Fur-mediated transcriptional repression is decreased resulting in multiple physiologic changes in the bacteria, including a switch to planktonic growth [45,91]. Planktonic bacteria are known to be more susceptible to certain antibiotics, indicating a possible mechanism of iron chelation-induced sensitization to antibiotics. Additional study is needed in order to fully elucidate how altering bacterial iron metabolism can affect antibiotic susceptibility.
A final aspect that must be fully addressed is the potential for toxicity with the use of iron chelating and gallium-based compounds. The polyvalent cations that bind with high affinity to these compounds are essential for multiple host physiological processes, raising the possibility that altering their concentration/metabolism could have adverse effects on host cells and tissues. Although some iron chelators have been approved for clinical use in the treatment of non-infectious indications, toxicity associated with iron chelation therapy has been described [92,93]. Future study is needed in order to characterize the potential toxicity of different iron chelating and gallium compounds at concentrations used for the treatment of bacterial infections.
8. Conclusions and Future Directions
The absolute requirement for iron in multiple essential bacterial processes of many pathogenic bacteria has raised interest in the use of iron chelators and gallium compounds as antimicrobial therapies over the previous decade. The majority of studies assessing these compounds have characterized their antimicrobial activity in vitro. Although multiple compounds have demonstrated promising results against some of the most important multidrug resistant strains, studies characterizing the antimicrobial activity of these compounds under physiological conditions are needed in order to fully evaluate their potential use as therapeutics. In-vivo studies in animal models are of special interest as they account for factors such as biodistribution and the presence of host factors, aspects which cannot be adequately addressed in most in vitro assays. Studies describing potential synergistic effects of iron chelators and gallium compounds when used with existing antibiotics indicate that combination therapy may be an effective therapeutic approach. Additional work is needed to understand the mechanisms by which iron chelators and gallium potentiate the activity of antibiotics.
Author Contributions
V.V. and M.J.M. designed, wrote and approved the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants MPY 380/18 from the Instituto de Salud Carlos III (ISCIII) awarded to M.J.M. V.V. is supported by the Río Hortega Program from the Instituto de Salud Carlos III.
Conflicts of Interest
M.J.M. is a founder and shareholder in the biotechnology company Vaxdyn, S.L. Vaxdyn played no role in the present study. No other competing interest is declared.
References
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Karakonstantis, S.; Kritsotakis, E.I.; Gikas, A. Pandrug-resistant Gram-negative bacteria: A systematic review of current epidemiology, prognosis and treatment options. J. Antimicrob. Chemother. 2019, 75, 271–282. [Google Scholar] [CrossRef]
- Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014. Available online: http://amrreview.org/sites/default/files/ (accessed on 9 February 2021).
- Theuretzbacher, U.; Bush, K.; Harbarth, S.; Paul, M.; Rex, J.H.; Tacconelli, E.; Thwaites, G.E. Critical analysis of antibacterial agents in clinical development. Nat. Rev. Genet. 2020, 18, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics—A pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Foley, T.L.; Simeonov, A. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discov. 2012, 7, 831–847. [Google Scholar] [CrossRef]
- Negash, K.H.; Norris, J.K.; Hodgkinson, J.T. Siderophore—Antibiotic conjugate design: New drugs for bad bugs? Molecules 2019, 24, 3314. [Google Scholar] [CrossRef]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.; Leibman, A. Lactoferrin and transferrin: A comparative study. Biochim. Biophys. Acta Protein Struct. 1972, 257, 314–323. [Google Scholar] [CrossRef]
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J. Biol. Chem. 1978, 253, 1930–1937. [Google Scholar] [CrossRef]
- Mazurier, J.; Spik, G. Comparative study of the iron-binding properties of human transferrins: I. Complete and sequential iron saturation and desaturation of the lactotransferrin. Biochim. Biophys. Acta Gen. Subj. 1980, 629, 399–408. [Google Scholar] [CrossRef]
- Huang, W.; Wilks, A. Extracellular heme uptake and the challenge of bacterial cell membranes. Annu. Rev. Biochem. 2017, 86, 799–823. [Google Scholar] [CrossRef]
- Palmer, L.D.; Skaar, E.P. Transition metals and virulence in bacteria. Annu. Rev. Genet. 2016, 50, 67–91. [Google Scholar] [CrossRef]
- Richard, K.L.; Kelley, B.R.; Johnson, J.G. Heme uptake and utilization by gram-negative bacterial pathogens. Front. Cell. Infect. Microbiol. 2019, 9, 81. [Google Scholar] [CrossRef]
- Imperi, F.; Tiburzi, F.; Visca, P. Molecular basis of pyoverdine siderophore recycling in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2009, 106, 20440–20445. [Google Scholar] [CrossRef]
- Wandersman, C.; Delepelaire, P. Bacterial iron sources: From siderophores to hemophores. Annu. Rev. Microbiol. 2004, 58, 611–647. [Google Scholar] [CrossRef]
- Lau, C.K.Y.; Krewulak, K.D.; Vogel, H.J. Bacterial ferrous iron transport: The feo system. FEMS Microbiol. Rev. 2016, 40, 273–298. [Google Scholar] [CrossRef]
- Zhou, T.; Winkelmann, G.; Dai, Z.-Y.; Hider, R.C. Design of clinically useful macromolecular iron chelators. J. Pharm. Pharmacol. 2011, 63, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.D.; Hider, R.C. Design of clinically useful iron (III)-selective chelators. Med. Res. Rev. 2001, 22, 26–64. [Google Scholar] [CrossRef]
- Brock, J.H.; Licéaga, J.; Kontoghiorghes, G.J. The effect of synthetic iron chelators on bacterial growth in human serum. FEMS Microbiol. Immunol. 1988, 1, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Gordeuk Vr Fau-Thuma, P.E.; Thuma Pe Fau-Theanacho, E.N.; Theanacho En Fau-Spira, D.T.; Spira Dt Fau-Hider, R.C.; Hider Rc Fau-Peto, T.E.; Peto Te Fau-Brittenham, G.M.; Brittenham, G.M. The antimalarial effect of iron chelators: Studies in animal models and in humans with mild falciparum malaria. J. Inorg. Biochem. 1992, 47, 267–277. [Google Scholar] [CrossRef]
- Gokarn, K.; Pal, R.B. Activity of siderophores against drug-resistant Gram-positive and Gram-negative bacteria. Infect. Drug Resist. 2018, 11, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Corey, B.W.; Si, Y.; Craft, D.W.; Zurawski, D.V. Antibacterial activities of iron chelators against common nosocomial pathogens. Antimicrob. Agents Chemother. 2012, 56, 5419–5421. [Google Scholar] [CrossRef] [PubMed]
- Moreau-Marquis, S.; O’Toole, G.A.; Stanton, B.A. Tobramycin and FDA-Approved Iron Chelators Eliminate Pseudomonas aeruginosa biofilms on cystic fibrosis cells. Am. J. Respir. Cell Mol. Biol. 2009, 41, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Banin, E.; Lozinski, A.; Brady, K.M.; Berenshtein, E.; Butterfield, P.W.; Moshe, M.; Chevion, M.; Greenberg, E.P. The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proc. Natl. Acad. Sci. USA 2008, 105, 16761–16766. [Google Scholar] [CrossRef] [PubMed]
- Carlson-Banning, K.M.; Chou, A.; Liu, Z.; Hamill, R.J.; Song, Y.; Zechiedrich, L. Toward repurposing ciclopirox as an antibiotic against drug-resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae. PLoS ONE 2013, 8, e69646. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Parquet, M.; Savage, K.A.; Allan, D.S.; Ang, M.T.C.; Chen, W.; Logan, S.M.; Holbein, B.E. Antibiotic-resistant Acinetobacter baumannii is susceptible to the novel iron-sequestering anti-infective dibi in vitro and in experimental pneumonia in mice. Antimicrob. Agents Chemother. 2019, 63, 00855-19. [Google Scholar] [CrossRef]
- López-Rojas, R.; García-Quintanilla, M.; Labrador-Herrera, G.; Pachón, J.; McConnell, M.J. Impaired growth under iron-limiting conditions associated with the acquisition of colistin resistance in Acinetobacter baumannii. Int. J. Antimicrob. Agents 2016, 47, 473–477. [Google Scholar] [CrossRef]
- Qiu, D.-H.; Huang, Z.-L.; Zhou, T.; Shen, C.; Hider, R.C. In vitro inhibition of bacterial growth by iron chelators. FEMS Microbiol. Lett. 2010, 314, 107–111. [Google Scholar] [CrossRef]
- Sobke, A.; Klinger, M.; Hermann, B.; Sachse, S.; Nietzsche, S.; Makarewicz, O.; Keller, P.M.; Pfister, W.; Straube, E. The Urinary Antibiotic 5-Nitro-8-hydroxyquinoline (Nitroxoline) reduces the formation and induces the dispersal of Pseudomonas aeruginosa biofilms by chelation of iron and zinc. Antimicrob. Agents Chemother. 2012, 56, 6021–6025. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Parquet, M.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B.E. Novel iron-chelator DIBI inhibits Staphylococcus aureus growth, suppresses experimental MRSA infection in mice and enhances the activities of diverse antibiotics in vitro. Front. Microbiol. 2018, 9, 1811. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.S.; Del Carmen Parquet, M.; Savage, K.A.; Holbein, B.E. Iron sequestrant DIBI, a Potential alternative for nares decolonization of methicillin-resistant Staphylococcus aureus, Is anti-infective and inhibitory for mupirocin-resistant isolates. Antimicrob. Agents Chemother. 2020, 64, 64. [Google Scholar] [CrossRef]
- Luo, G.; Spellberg, B.; Gebremariam, T.; Lee, H.; Xiong, Y.Q.; French, S.W.; Bayer, A.; Ibrahim, A.S. Combination therapy with iron chelation and vancomycin in treating murine staphylococcemia. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lin, Y.; Lu, Q.; Li, F.; Yu, J.; Wang, Z.; He, Y.; Song, C. In vitro and in vivo activity of EDTA and antibacterial agents against the biofilm of mucoid Pseudomonas aeruginosa. Infection 2016, 45, 23–31. [Google Scholar] [CrossRef] [PubMed]
- O’May, C.Y.; Sanderson, K.; Roddam, L.F.; Kirov, S.M.; Reid, D.W. Iron-binding compounds impair Pseudomonas aeruginosa biofilm formation, especially under anaerobic conditions. J. Med. Microbiol. 2009, 58, 765–773. [Google Scholar] [CrossRef]
- Llamas, M.A.; Sparrius, M.; Kloet, R.; Jiménez, C.R.; Vandenbroucke-Grauls, C.; Bitter, W. The heterologous siderophores ferrioxamine b and ferrichrome activate signaling pathways in Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Cuív, P.O.; Keogh, D.; Clarke, P.; O’Connell, M. FoxB of Pseudomonas aeruginosa functions in the utilization of the xenosiderophores ferrichrome, ferrioxamine b, and schizokinen: Evidence for transport redundancy at the inner membrane. J. Bacteriol. 2006, 189, 284–287. [Google Scholar] [CrossRef]
- Sebulsky, M.T.; Hohnstein, D.; Hunter, M.D.; Heinrichs, D.E. Identification and characterization of a membrane permease involved in iron-hydroxamate transport in Staphylococcus aureus. J. Bacteriol. 2000, 182, 4394–4400. [Google Scholar] [CrossRef]
- Clarke, T.E.; Braun, V.; Winkelmann, G.; Tari, L.W.; Vogel, H.J. X-ray crystallographic structures of the Escherichia coli periplasmic protein fhud bound to hydroxamate-type siderophores and the antibiotic albomycin. J. Biol. Chem. 2002, 277, 13966–13972. [Google Scholar] [CrossRef]
- Visca, P.; Bonchi, C.; Minandri, F.; Frangipani, E.; Imperi, F. The dual personality of iron chelators: Growth inhibitors or promoters? Antimicrob. Agents Chemother. 2013, 57, 2432–2433. [Google Scholar] [CrossRef]
- De Léséleuc, L.; Harris, G.; KuoLee, R.; Chen, W. In vitro and in vivo biological activities of iron chelators and gallium nitrate against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 5397–5400. [Google Scholar] [CrossRef]
- Huayhuaz, J.A.A.; Vitorino, H.A.; Campos, O.S.; Serrano, S.H.P.; Kaneko, T.M.; Espósito, B.P. Desferrioxamine and desferrioxamine-caffeine as carriers of aluminum and gallium to microbes via the Trojan Horse Effect. J. Trace Elements Med. Biol. 2017, 41, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Pippard, M.J.; Jackson, M.J.; Hoffman, K.; Petrou, M.; Modell, C.B. Iron chelation using subcutaneous infusions of diethylene triamine penta-acetic acid (DTPA). Scand. J. Haematol. 2009, 36, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Banin, E.; Brady, K.M.; Greenberg, E.P. Chelator-induced dispersal and killing of Pseudomonas aeruginosa cells in a biofilm. Appl. Environ. Microbiol. 2006, 72, 2064–2069. [Google Scholar] [CrossRef]
- Poole, K.; Krebes, K.; McNally, C.; Neshat, S. Multiple antibiotic resistance in Pseudomonas aeruginosa: Evidence for involvement of an efflux operon. J. Bacteriol. 1993, 175, 7363–7372. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; Molin, S. Synergistic activities of an efflux pump inhibitor and iron chelators against Pseudomonas aeruginosa growth and biofilm formation. Antimicrob. Agents Chemother. 2010, 54, 3960–3963. [Google Scholar] [CrossRef]
- Cohen, A.R. New Advances in Iron Chelation Therapy. In Hematology. American Society of Hematology. Education Program; ASH: Washington, DC, USA, 2006. [Google Scholar]
- Kim, C.-M.; Shin, S.-H. Effect of iron-chelator deferiprone on the in vitro growth of staphylococci. J. Korean Med. Sci. 2009, 24, 289–295. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. Iron mobilization from transferrin and non-transferrin-bound-iron by deferiprone. Implications in the treatment of thalassemia, anemia of chronic disease, cancer and other conditions. Hemoglobin 2006, 30, 183–200. [Google Scholar] [CrossRef]
- Niewerth, M.; Kunze, D.; Seibold, M.; Schaller, M.; Korting, H.C.; Hube, B. Ciclopirox olamine treatment affects the expression pattern of Candida albicans genes encoding virulence factors, iron metabolism proteins, and drug resistance factors. Antimicrob. Agents Chemother. 2003, 47, 1805–1817. [Google Scholar] [CrossRef]
- Sigle, H.-C.; Thewes, S.; Niewerth, M.; Korting, H.C.; Schäfer-Korting, M.; Hube, B. Oxygen accessibility and iron levels are critical factors for the antifungal action of ciclopirox against Candida albicans. J. Antimicrob. Chemother. 2005, 55, 663–673. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Liu, M.-S.; Osamah, A.R.; Kong, X.-L.; Alsam, S.; Battah, S.; Xie, Y.-Y.; Hider, R.C.; Zhou, T. Hexadentate 3-hydroxypyridin-4-ones with high iron (III) affinity: Design, synthesis and inhibition on methicillin resistant Staphylococcus aureus and Pseudomonas strains. Eur. J. Med. Chem. 2015, 94, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-J.; Zhang, M.-X.; Hider, R.C.; Zhou, T. In vitro antimicrobial activity of hydroxypyridinone hexadentate-based dendrimeric chelators alone and in combination with norfloxacin. FEMS Microbiol. Lett. 2014, 355, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Ang, M.T.C.; Gumbau-Brisa, R.; Allan, D.S.; McDonald, R.; Ferguson, M.J.; Holbein, B.E.; Bierenstiel, M. DIBI, a 3-hydroxypyridin-4-one chelator iron-binding polymer with enhanced antimicrobial activity. MedChemComm 2018, 9, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Fokam, D.; Dickson, K.; Kamali, K.; Holbein, B.; Colp, P.; Stueck, A.; Zhou, J.; Lehmann, C. Iron chelation in murine models of systemic inflammation induced by gram-positive and gram-negative toxins. Antibiotics 2020, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Aali, M.; Zhou, J.; Holbein, B. Comparison of Treatment effects of different iron chelators in experimental models of sepsis. Life 2021, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Parsek, M.R.; Greenberg, E.P.; Welsh, M.J. A component of innate immunity prevents bacterial biofilm development. Nature 2002, 417, 552–555. [Google Scholar] [CrossRef]
- Ardehali, R.; Shi, L.; Janatova, J.; Mohammad, S.F.; Burns, G.L. The effect of apo-transferrin on bacterial adhesion to biomaterials. Artif. Organs 2002, 26, 512–520. [Google Scholar] [CrossRef]
- Ammons, M.C.B.; Ward, L.S.; Dowd, S.; James, G.A. Combined treatment of Pseudomonas aeruginosa biofilm with lactoferrin and xylitol inhibits the ability of bacteria to respond to damage resulting from lactoferrin iron chelation. Int. J. Antimicrob. Agents 2011, 37, 316–323. [Google Scholar] [CrossRef][Green Version]
- Lin, L.; Pantapalangkoor, P.; Tan, B.; Bruhn, K.W.; Ho, T.; Nielsen, T.; Skaar, E.P.; Zhang, Y.; Bai, R.; Wang, A.; et al. Transferrin iron starvation therapy for lethal bacterial and fungal infections. J. Infect. Dis. 2014, 210, 254–264. [Google Scholar] [CrossRef]
- Luna, B.M.; Ershova, K.; Yan, J.; Ulhaq, A.; Nielsen, T.B.; Hsieh, S.; Pantapalangkoor, P.; Vanscoy, B.; Ambrose, P.; Rudin, S.; et al. Adjunctive transferrin to reduce the emergence of antibiotic resistance in Gram-negative bacteria. J. Antimicrob. Chemother. 2019, 74, 2631–2639. [Google Scholar] [CrossRef]
- Jeong, B.C.; Hawes, C.; Bonthrone, K.M.; Macaskie, L.E. Localization of enzymically enhanced heavy metal accumulation by Citrobacter sp. and metal accumulation in vitro by liposomes containing entrapped enzyme. Microbiology 1997, 143, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Ahire, J.J.; Dicks, L.M.T. 2,3-Dihydroxybenzoic Acid-containing nanofiber wound dressings inhibit biofilm formation by Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 2098–2104. [Google Scholar] [CrossRef]
- El-Gendy, N.; Qian, J.; Eshelman, K.; Rivera, M.; Berkland, C. Antibiotic activity of iron-sequestering polymers. Biomacromolecules 2015, 16, 1480–1488. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. A record Of 1320 suspect, deferasirox-related, patient deaths reported in 2009: Insufficient toxicity testing, low efficacy and lack of transparency may endanger the lives of iron loaded patients. Hemoglobin 2011, 35, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Crooks, P.A.; Wei, X.; Leon, D.J. Toxicity of dipyridyl compounds and related compounds. Crit. Rev. Toxicol. 2004, 34, 447–460. [Google Scholar] [CrossRef]
- Antunes, L.C.S.; Imperi, F.; Minandri, F.; Visca, P. In vitro and in vivo antimicrobial activities of gallium nitrate against multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 5961–5970. [Google Scholar] [CrossRef] [PubMed]
- Carretero-Ledesma, M.; García-Quintanilla, M.; Martín-Peña, R.; Pulido, M.R.; Pachón, J.; McConnell, M.J. Phenotypic changes associated with Colistin resistance due to Lipopolysaccharide loss in Acinetobacter baumannii. Virulence 2018, 9, 930–942. [Google Scholar] [CrossRef]
- Pelletier, C.; Prognon, P.; Bourlioux, P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob. Agents Chemother. 1995, 39, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, L.R. Mechanisms of therapeutic activity for gallium. Pharmacol. Rev. 1998, 50, 665–682. [Google Scholar]
- Chitambar, C.R. Gallium and its competing roles with iron in biological systems. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, B.; Xie, Y.; Wang, H.; Wang, R.; Xia, W.; Li, H.; Sun, H. Combination of gallium(iii) with acetate for combating antibiotic resistant Pseudomonas aeruginosa. Chem. Sci. 2019, 10, 6099–6106. [Google Scholar] [CrossRef] [PubMed]
- Runci, F.; Bonchi, C.; Frangipani, E.; Visaggio, D.; Visca, P. Acinetobacter baumannii Biofilm formation in human serum and disruption by gallium. Antimicrob. Agents Chemother. 2016, 61, e01563-16. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.C.; Sayood, K.; Olmsted, S.B.; Blanchard, C.E.; Hinrichs, S.; Russell, D.; Dunman, P.M. Characterization of the Acinetobacter baumannii growth phase-dependent and serum responsive transcriptomes. FEMS Immunol. Med. Microbiol. 2012, 64, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.D.; Heymann, J.J.; Mehta, A.; Roulhac, P.L.; Anderson, D.S.; Nowalk, A.J.; Adhikari, P.; Mietzner, T.A.; Fitzgerald, M.C.; Crumbliss, A.L. Ga3+ as a mechanistic probe in Fe3+ transport: Characterization of Ga3+ interaction with FbpA. JBIC J. Biol. Inorg. Chem. 2008, 13, 887–898. [Google Scholar] [CrossRef]
- Frangipani, E.; Bonchi, C.; Minandri, F.; Imperi, F.; Visca, P. Pyochelin potentiates the inhibitory activity of gallium on Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 5572–5575. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Tennent, D.J.; Chang, D.; Wenke, J.C.; Sanchez, C.J. An In Vitro Comparison of PMMA and calcium sulfate as carriers for the local delivery of gallium (III) nitrate to staphylococcal infected surgical sites. BioMed Res. Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-R.; Britigan, B.E.; Narayanasamy, P. Dual Inhibition of Klebsiella pneumoniae and Pseudomonas aeruginosa iron metabolism using gallium porphyrin and gallium nitrate. ACS Infect. Dis. 2019, 5, 1559–1569. [Google Scholar] [CrossRef]
- Hijazi, S.; Visaggio, D.; Pirolo, M.; Frangipani, E.; Bernstein, L.; Visca, P. Antimicrobial activity of gallium compounds on ESKAPE Pathogens. Front. Cell. Infect. Microbiol. 2018, 8, 316. [Google Scholar] [CrossRef]
- Arivett, B.A.; Fiester, S.E.; Ohneck, E.J.; Penwell, W.F.; Kaufman, C.M.; Relich, R.F.; Actis, L.A. Antimicrobial activity of gallium protoporphyrin IX against Acinetobacter baumannii strains displaying different antibiotic resistance phenotypes. Antimicrob. Agents Chemother. 2015, 59, 7657–7665. [Google Scholar] [CrossRef]
- Thompson, M.G.; Truong-Le, V.; Alamneh, Y.A.; Black, C.C.; Anderl, J.; Honnold, C.L.; Pavlicek, R.L.; Abu-Taleb, R.; Wise, M.C.; Hall, E.R.; et al. Evaluation of gallium citrate formulations against a multidrug-resistant strain of Klebsiella pneumoniae in a murine wound model of infection. Antimicrob. Agents Chemother. 2015, 59, 6484–6493. [Google Scholar] [CrossRef]
- Baldoni, D.; Steinhuber, A.; Zimmerli, W.; Trampuz, A. In vitro activity of gallium maltolate against staphylococci in logarithmic, stationary, and biofilm growth phases: Comparison of conventional and calorimetric susceptibility testing methods. Antimicrob. Agents Chemother. 2009, 54, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, S.; Visca, P.; Frangipani, E. Gallium-protoporphyrin IX inhibits Pseudomonas aeruginosa growth by targeting cytochromes. Front. Cell. Infect. Microbiol. 2017, 7, 12. [Google Scholar] [CrossRef]
- Stojiljkovic, I.; Kumar, V.; Srinivasan, N. Non-iron metalloporphyrins: Potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 1999, 31, 429–442. [Google Scholar] [CrossRef]
- Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B.E.; Singh, P.K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Investig. 2007, 117, 877–888. [Google Scholar] [CrossRef]
- Goss, C.H.; Kaneko, Y.; Khuu, L.; Anderson, G.D.; Ravishankar, S.; Aitken, M.L.; Lechtzin, N.; Zhou, G.; Czyz, D.M.; McLean, K.; et al. Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci. Transl. Med. 2018, 10, eaat7520. [Google Scholar] [CrossRef]
- Richter, K.; Ramezanpour, M.; Thomas, N.; Prestidge, C.A.; Wormald, P.; Vreugde, S. Mind “De GaPP”: In vitro efficacy of deferiprone and gallium-protoporphyrin against Staphylococcus aureus biofilms. Int. Forum Allergy Rhinol. 2016, 6, 737–743. [Google Scholar] [CrossRef]
- Richter, K.; Thomas, N.; Claeys, J.; McGuane, J.; Prestidge, C.A.; Coenye, T.; Wormald, P.-J.; Vreugde, S. A Topical hydrogel with deferiprone and gallium-protoporphyrin targets bacterial iron metabolism and has antibiofilm activity. Antimicrob. Agents Chemother. 2017, 61, e00481-17. [Google Scholar] [CrossRef] [PubMed]
- Escolar, L.; Pérez-Martín, J.; De Lorenzo, V. Opening the iron box: Transcriptional metalloregulation by the fur protein. J. Bacteriol. 1999, 181, 6223–6229. [Google Scholar] [CrossRef]
- Hancock, V.; Dahl, M.; Klemm, P. Abolition of biofilm formation in urinary tract Escherichia coli and Klebsiella isolates by metal interference through competition for fur. Appl. Environ. Microbiol. 2010, 76, 3836–3841. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E. Clinical application of deferasirox: Practical patient management. Am. J. Hematol. 2008, 83, 398–402. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).