
Anti-Cancer Effects and Tumor Marker Role of Glutathione S-Transferase Mu 5 in Human Bladder Cancer


Abstract
:1. Introduction
2. Results
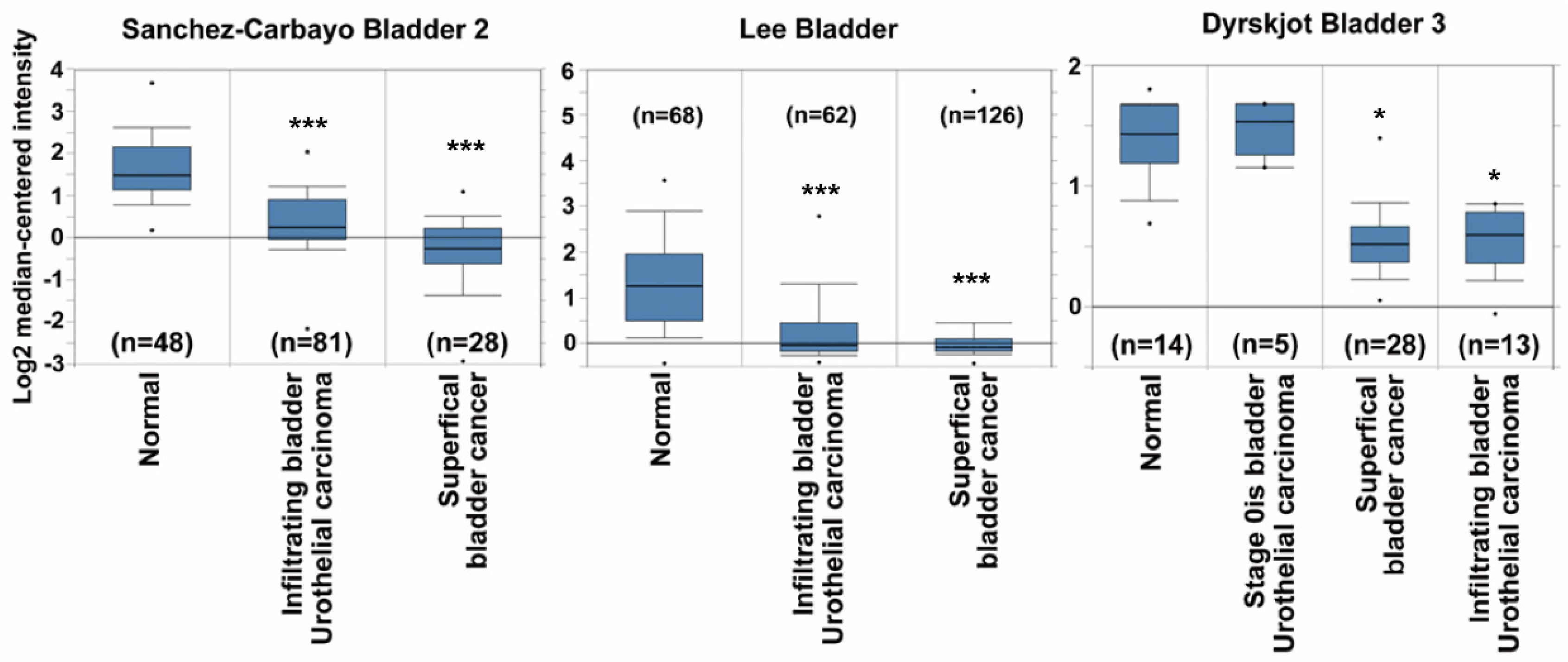
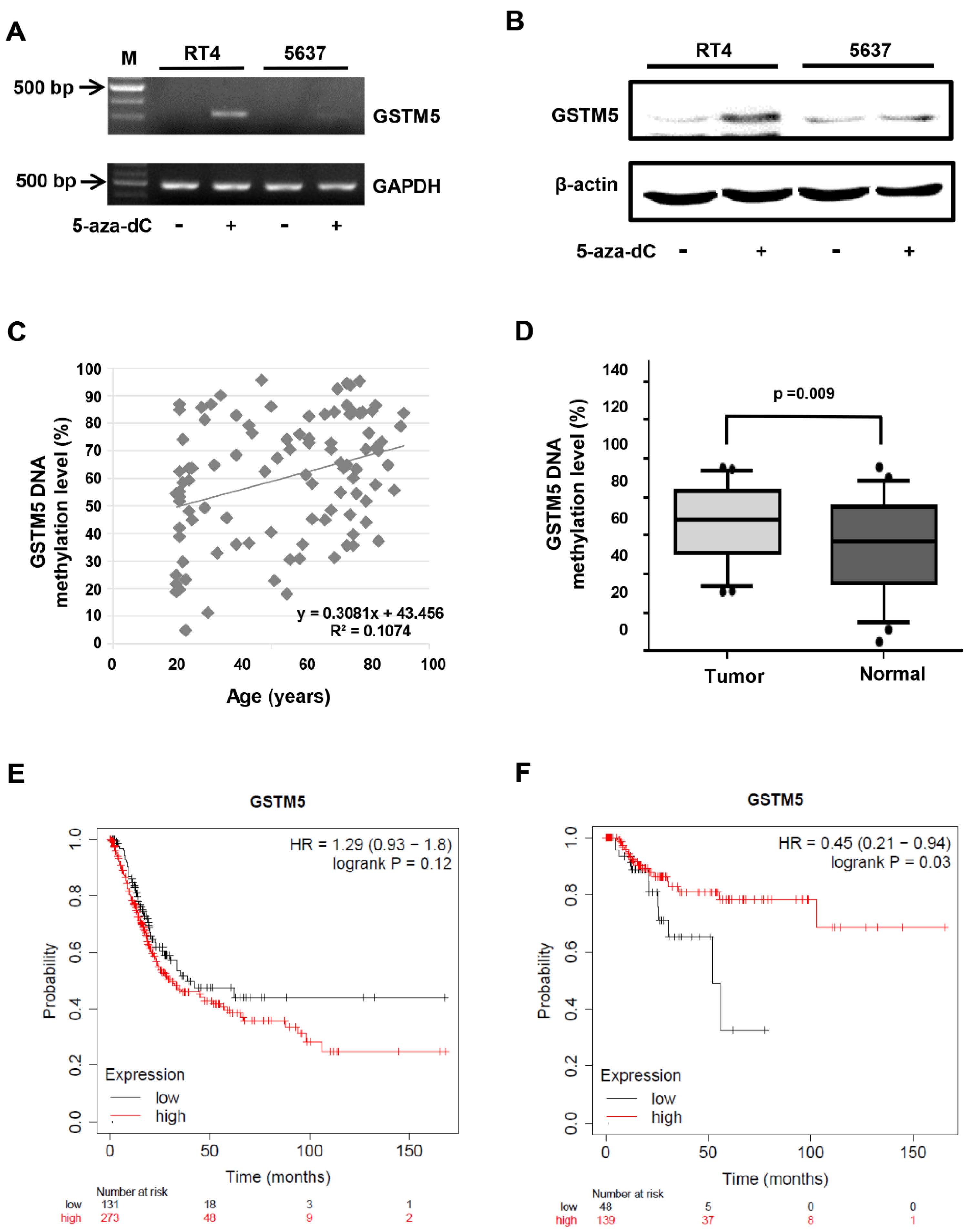
2.1. GSTM5 mRNA Expression Is Downregulated in Bladder Cancer Tissues
2.2. DNA Methylation Level of the GSTM5 Gene Promoter Is Increased in Bladder Cancer Tissues
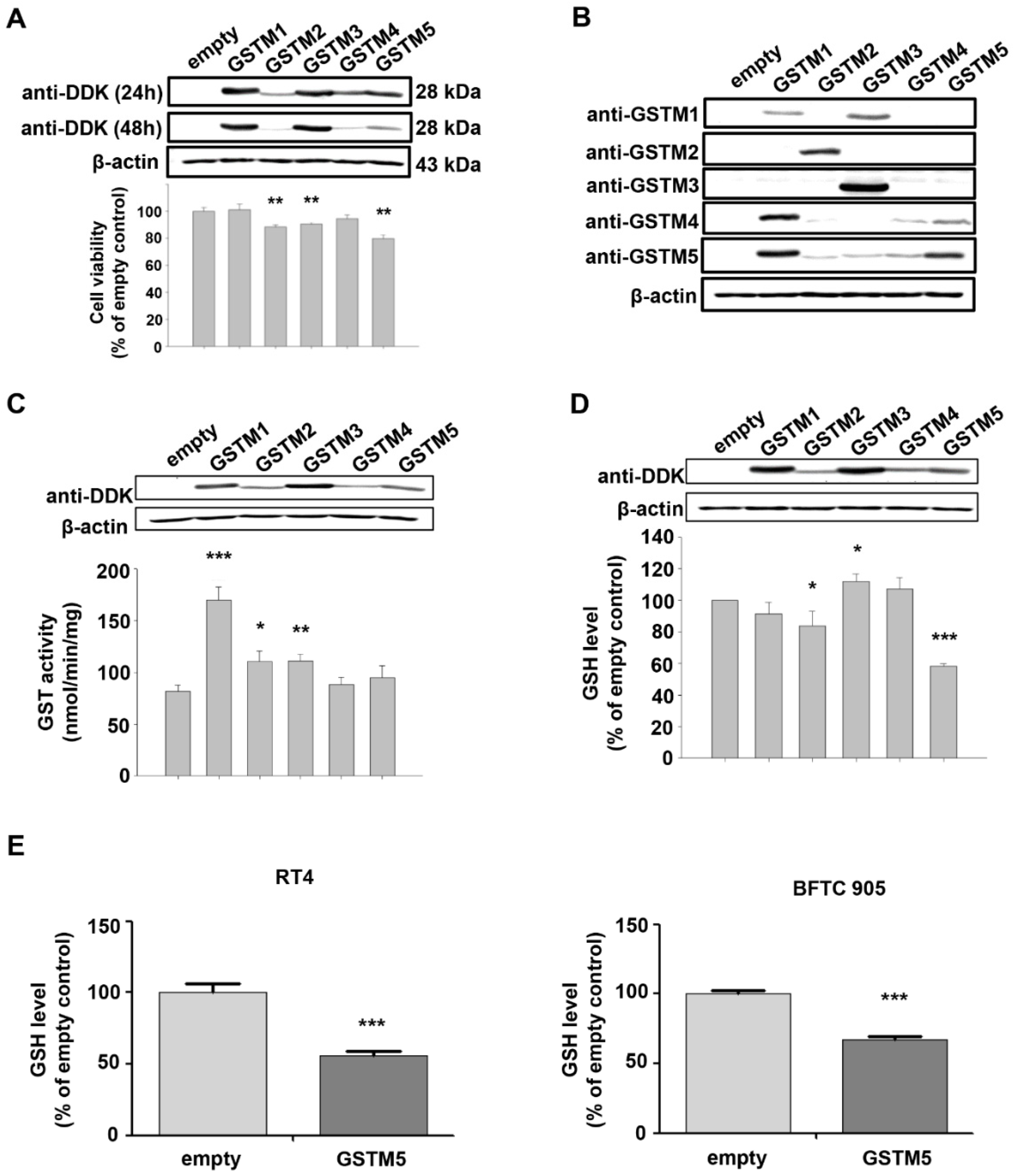
2.3. Biological Effects of GSTM1-5 Overexpression in Bladder Cancer Cells
2.4. GSTM5 Overexpression Decreases Intracellular GSH Levels and Suppresses the Proliferation and Migration of Bladder Cancer Cells
2.5. GSH Reverses GSTM5-Inhibited Cell Proliferation and Migration
2.6. BSO Administration Decreases Intracellular GSH and Suppresses the Proliferation and Migration of 5637 Bladder Cancer Cells
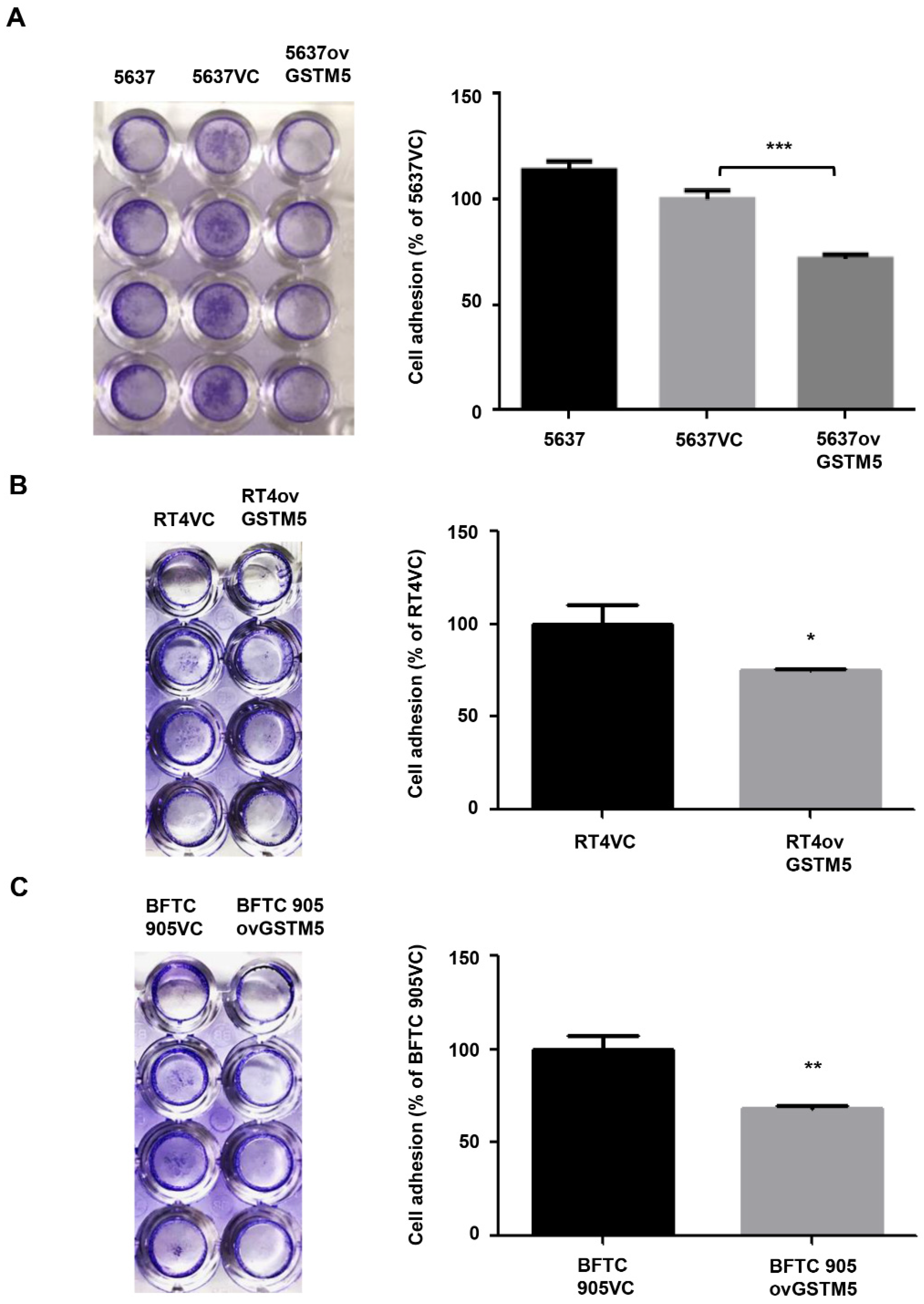
2.7. Cell Adhesion Capacity Is Decreased in GSTM5-Overexpressing Cells
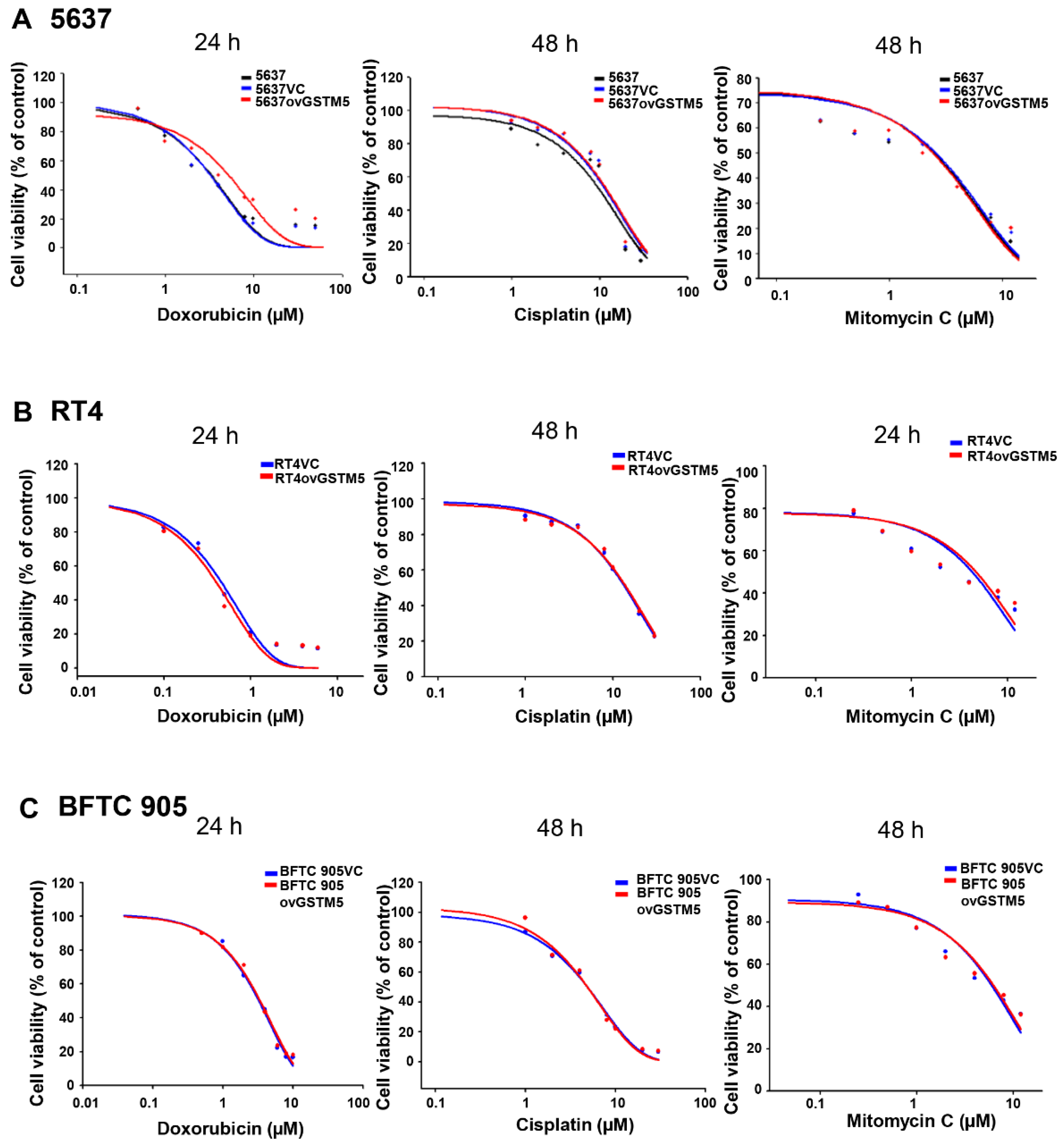
2.8. Effects of GSTM5 Overexpression on the Cytotoxic Sensitivity of Doxorubicin, Cisplatin and Mitomycin C
3. Discussion
4. Materials and Methods
4.1. Extraction of Genomic DNA from Human Bladder Tumors, Human Urine Pellets and Cultured Cell Lines
4.2. Bisulfite Conversion of Genomic DNA and Analysis of the DNA Methylation Level in Human Samples
4.3. Cell Culture and RT–PCR
4.4. Western Blot
4.5. GSTMs Overexpression in 5637 Cells
4.6. GSTM5 Overexpression in RT4 and BFTC 905 Cells
4.7. GST Activity Assay
4.8. Cellular GSH Quantification
4.9. Cell Proliferation Assay
4.10. Colony Formation Assay
4.11. Migration Assay
4.12. Wound Healing Assay
4.13. Cell Adhesion Assay
4.14. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Stenehjem, D.D.; Tran, D.; Nkrumah, M.A.; Gupta, S. PD1/PDL1 inhibitors for the treatment of advanced urothelial bladder cancer. Onco Targets Ther. 2018, 11, 5973–5989. [Google Scholar] [CrossRef] [Green Version]
- Lower, G.M., Jr. Concepts in causality: Chemically induced human urinary bladder cancer. Cancer 1982, 49, 1056–1066. [Google Scholar] [CrossRef] [Green Version]
- Brennan, P.; Bogillot, O.; Cordier, S.; Greiser, E.; Schill, W.; Vineis, P.; Lopez-Abente, G.; Tzonou, A.; Chang-Claude, J.; Bolm-Audorff, U.; et al. Cigarette smoking and bladder cancer in men: A pooled analysis of 11 case-control studies. Int. J. Cancer 2000, 86, 289–294. [Google Scholar] [CrossRef]
- Straif, K.; Benbrahim-Tallaa, L.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part C: Metals, arsenic, dusts, and fibres. Lancet Oncol. 2009, 10, 453–454. [Google Scholar] [CrossRef]
- Jou, Y.C.; Wang, S.C.; Dai, Y.C.; Chen, S.Y.; Shen, C.H.; Lee, Y.R.; Chen, L.C.; Liu, Y.W. Gene expression and DNA methylation regulation of arsenic in mouse bladder tissues and in human urothelial cells. Oncol. Rep. 2019, 42, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Strange, R.C. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 2000, 61, 154–166. [Google Scholar] [CrossRef]
- Engel, L.S.; Taioli, E.; Pfeiffer, R.; Garcia-Closas, M.; Marcus, P.M.; Lan, Q.; Boffetta, P.; Vineis, P.; Autrup, H.; Bell, D.A.; et al. Pooled analysis and meta-analysis of glutathione S-transferase M1 and bladder cancer: A HuGE review. Am. J. Epidemiol. 2002, 156, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.W.; Song, P.H.; Ha, Y.S.; Kim, W.T.; Kim, Y.J.; Yun, S.J.; Lee, S.C.; Choi, Y.H.; Moon, S.K.; Kim, W.J. Glutathione S-transferase M1 and T1 polymorphisms: Susceptibility and outcomes in muscle invasive bladder cancer patients. Eur. J. Cancer 2013, 49, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Matic, M.; Pekmezovic, T.; Djukic, T.; Mimic-Oka, J.; Dragicevic, D.; Krivic, B.; Suvakov, S.; Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Tulic, C.; et al. GSTA1, GSTM1, GSTP1, and GSTT1 polymorphisms and susceptibility to smoking-related bladder cancer: A case-control study. Urol. Oncol. 2013, 31, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.J.; Dai, Y.C.; Lin, Y.L.; Chen, Y.Y.; Lin, W.H.; Chan, H.L.; Liu, Y.W. Downregulation of glutathione S-transferase M1 protein in N-butyl-N-(4-hydroxybutyl)nitrosamine-induced mouse bladder carcinogenesis. Toxicol. Appl. Pharmacol. 2014, 279, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R.; Vorachek, W.R.; Xu, S.J.; Berger, R.; Hart, I.; Vannais, D.; Patterson, D. Identification of class-mu glutathione transferase genes GSTM1-GSTM5 on human chromosome 1p13. Am. J. Hum. Genet. 1993, 53, 220–233. [Google Scholar]
- Bhattacharjee, P.; Paul, S.; Banerjee, M.; Patra, D.; Banerjee, P.; Ghoshal, N.; Bandyopadhyay, A.; Giri, A.K. Functional compensation of glutathione S-transferase M1 (GSTM1) null by another GST superfamily member, GSTM2. Sci. Rep. 2013, 3, 2704. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.D.; Fan, L.; Di, G.H.; Yuan, W.T.; Zheng, Y.; Huang, W.; Chen, A.X.; Yang, C.; Wu, J.; Shen, Z.Z.; et al. Genetic variants in GSTM3 gene within GSTM4-GSTM2-GSTM1-GSTM5-GSTM3 cluster influence breast cancer susceptibility depending on GSTM1. Breast Cancer Res. Treat. 2010, 121, 485–496. [Google Scholar] [CrossRef]
- Weng, M.W.; Hsiao, Y.M.; Chiou, H.L.; Yang, S.F.; Hsieh, Y.S.; Cheng, Y.W.; Yang, C.H.; Ko, J.L. Alleviation of benzo[a]pyrene-diolepoxide-DNA damage in human lung carcinoma by glutathione S-transferase M2. DNA Repair 2005, 4, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Sheu, G.T.; Wong, R.H.; Huang, C.Y.; Weng, M.W.; Lee, L.W.; Hsu, C.P.; Ko, J.L. Expression of glutathione S-transferase M2 in stage I/II non-small cell lung cancer and alleviation of DNA damage exposure to benzo[a]pyrene. Toxicol. Lett. 2010, 192, 316–323. [Google Scholar] [CrossRef]
- Tang, S.C.; Wu, M.F.; Wong, R.H.; Liu, Y.F.; Tang, L.C.; Lai, C.H.; Hsu, C.P.; Ko, J.L. Epigenetic mechanisms for silencing glutathione S-transferase m2 expression by hypermethylated specificity protein 1 binding in lung cancer. Cancer 2011, 117, 3209–3221. [Google Scholar] [CrossRef]
- Tang, S.C.; Wu, C.H.; Lai, C.H.; Sung, W.W.; Yang, W.J.; Tang, L.C.; Hsu, C.P.; Ko, J.L. Glutathione S-transferase mu2 suppresses cancer cell metastasis in non-small cell lung cancer. Mol. Cancer Res. MCR 2013, 11, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Ibragimova, I.; Dulaimi, E.; Slifker, M.J.; Chen, D.Y.; Uzzo, R.G.; Cairns, P. A global profile of gene promoter methylation in treatment-naive urothelial cancer. Epigenetics 2014, 9, 760–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.F.; Razvi, M.; Chen, H.; Washington, K.; Roessner, A.; Schneider-Stock, R.; El-Rifai, W. DNA hypermethylation regulates the expression of members of the Mu-class glutathione S-transferases and glutathione peroxidases in Barrett’s adenocarcinoma. Gut 2009, 58, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Huang, C.C.; Shen, C.H.; Lin, L.C.; Zhao, P.W.; Chen, S.Y.; Deng, Y.C.; Liu, Y.W. Gene Expression and DNA Methylation Status of Glutathione S-Transferase Mu1 and Mu5 in Urothelial Carcinoma. PLoS ONE 2016, 11, e0159102. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, F.; Wang, C.; Wang, C.; Tang, Y.; Jiang, Z. Glutathione S-transferase A1 mediates nicotine-induced lung cancer cell metastasis by promoting epithelial-mesenchymal transition. Exp. Ther Med. 2017, 14, 1783–1788. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001, 15, 19–21. [Google Scholar] [CrossRef]
- Carretero, J.; Obrador, E.; Anasagasti, M.J.; Martin, J.J.; Vidal-Vanaclocha, F.; Estrela, J.M. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin. Exp. Metastasis 1999, 17, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Chen, Y.; Xu, R.; He, Q.Y. Nrf2 mediates the resistance of human A549 and HepG2 cancer cells to boningmycin, a new antitumor antibiotic, in vitro through regulation of glutathione levels. Acta Pharm. Sin. 2018, 39, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Harbottle, A.; Daly, A.K.; Atherton, K.; Campbell, F.C. Role of glutathione S-transferase P1, P-glycoprotein and multidrug resistance-associated protein 1 in acquired doxorubicin resistance. Int J. Cancer 2001, 92, 777–783. [Google Scholar] [CrossRef]
- Sawers, L.; Ferguson, M.J.; Ihrig, B.R.; Young, H.C.; Chakravarty, P.; Wolf, C.R.; Smith, G. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 2014, 111, 1150–1158. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Yang, Y.; Lei, C.; Yang, F.; Liang, L.; Chen, C.; Xia, J.; Wang, K.; Tang, N. GSTZ1 deficiency promotes hepatocellular carcinoma proliferation via activation of the KEAP1/NRF2 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 438. [Google Scholar] [CrossRef]
- Sun, C.; Gu, Y.; Chen, G.; Du, Y. Bioinformatics Analysis of Stromal Molecular Signatures Associated with Breast and Prostate Cancer. J. Comput. Biol. 2019, 26, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lei, L.; Chi, Y.G.; Liu, L.B.; Yang, B.P. A comprehensive understanding of ovarian carcinoma survival prognosis by novel biomarkers. Eur. Rev. Med. Pharm. Sci. 2019, 23, 8257–8264. [Google Scholar] [CrossRef]
- Hentschel, A.E.; Nieuwenhuijzen, J.A.; Bosschieter, J.; Splunter, A.P.V.; Lissenberg-Witte, B.I.; Voorn, J.P.V.; Segerink, L.I.; Moorselaar, R.; Steenbergen, R.D.M. Comparative Analysis of Urine Fractions for Optimal Bladder Cancer Detection Using DNA Methylation Markers. Cancers 2020, 12, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoque, M.O.; Begum, S.; Topaloglu, O.; Chatterjee, A.; Rosenbaum, E.; Van Criekinge, W.; Westra, W.H.; Schoenberg, M.; Zahurak, M.; Goodman, S.N.; et al. Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J. Natl. Cancer Inst. 2006, 98, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakami, S.; Shiina, H.; Enokida, H.; Kawakami, T.; Kawamoto, K.; Hirata, H.; Tanaka, Y.; Kikuno, N.; Nakagawa, M.; Igawa, M.; et al. Combination analysis of hypermethylated Wnt-antagonist family genes as a novel epigenetic biomarker panel for bladder cancer detection. Clin. Cancer Res. 2006, 12, 2109–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Bladder Cancer | Healthy Individual |
|---|---|---|
| Age (min/max) | 28/92 | 20/81 |
| Mean of age | 73.16 | 35.42 |
| Sexuality (male/female) | 32/18 | 11/39 |
| Sample Group | Male | Female | p-Value |
|---|---|---|---|
| Group | |||
| Bladder cancer number (%) | 32 (74.42) | 18 (31.58) | <0.001 |
| Healthy individual number (%) | 11 (25.58) | 39 (68.42) | |
| All samples | |||
| Methylation level (%) | 61.17 ± 22.62 | 59.43 ± 22.32 | 0.700 |
| Three-Tier Classification of Methylation Level | Bladder Cancer Number (%) | Healthy Individual Number (%) | p-Value |
|---|---|---|---|
| Methylation levels (%) | 0.003 | ||
| Low (<30) | 0 (0) | 10 (20) | |
| Medium (30 ≦ x < 75) | 32 (64) | 28 (56) | |
| High (≧75) | 18 (36) | 12 (24) |
| Cancer Stage | Methylation Levels | p-Value | ||
|---|---|---|---|---|
| Low (<30) Number (%) | Medium (30 ≦ x < 75) Number (%) | High (≧75) Number (%) | ||
| Bladder status | 0.031 | |||
| Normal | 10 (100) | 28 (47.67) | 12 (40) | |
| Stage 0is | 0 (0) | 9 (15) | 2 (6.67) | |
| Superficial | 0 (0) | 17 (28.33) | 11 (36.67) | |
| Infiltrating | 0 (0) | 6 (10) | 5 (16.67) | |
| Total | 10 (100) | 60 (100) | 30 (100) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jou, Y.-C.; Wang, S.-C.; Dia, Y.-C.; Wang, S.-T.; Yu, M.-H.; Yang, H.-Y.; Chen, L.-C.; Shen, C.-H.; Liu, Y.-W. Anti-Cancer Effects and Tumor Marker Role of Glutathione S-Transferase Mu 5 in Human Bladder Cancer. Int. J. Mol. Sci. 2021, 22, 3056. https://doi.org/10.3390/ijms22063056
Jou Y-C, Wang S-C, Dia Y-C, Wang S-T, Yu M-H, Yang H-Y, Chen L-C, Shen C-H, Liu Y-W. Anti-Cancer Effects and Tumor Marker Role of Glutathione S-Transferase Mu 5 in Human Bladder Cancer. International Journal of Molecular Sciences. 2021; 22(6):3056. https://doi.org/10.3390/ijms22063056
Chicago/Turabian StyleJou, Yeong-Chin, Shou-Chieh Wang, Yuan-Chang Dia, Shou-Tsung Wang, Min-Hua Yu, Hsin-Yi Yang, Lei-Chin Chen, Cheng-Huang Shen, and Yi-Wen Liu. 2021. "Anti-Cancer Effects and Tumor Marker Role of Glutathione S-Transferase Mu 5 in Human Bladder Cancer" International Journal of Molecular Sciences 22, no. 6: 3056. https://doi.org/10.3390/ijms22063056