
Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
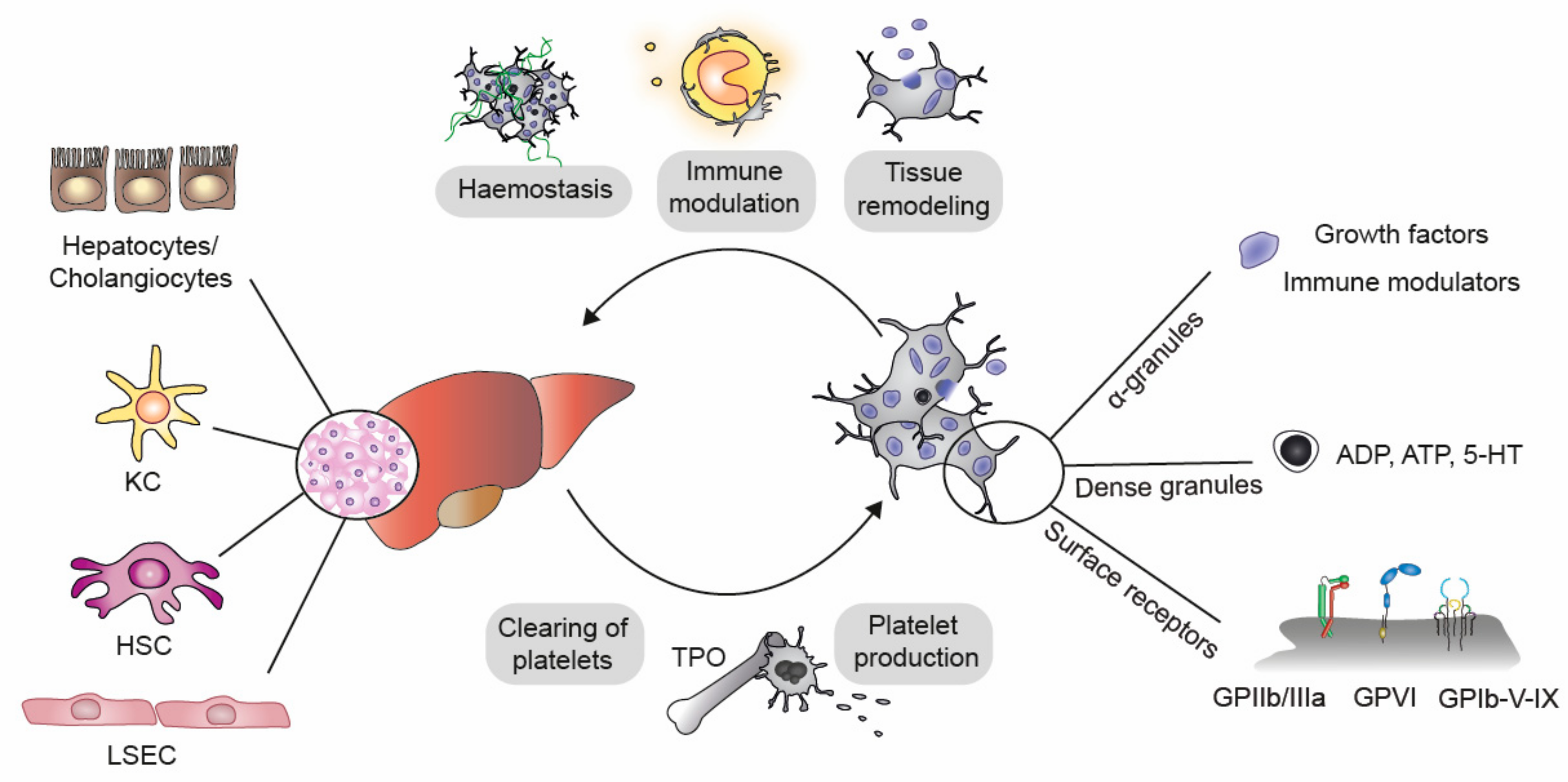
2. Mechanisms of Interaction between Platelets and Liver
3. Liver Diseases and Their Impact on Platelet Count
4. Hepatic Steatosis (NAFLD)
4.1. Procoagulatory and Prothrombotic Events at the Onset of Fatty Liver Diseases
4.2. The Role of Platelet-Derived Molecules in Fatty Liver Diseases
4.3. Other Factors Associated with Fatty Liver Disease
5. Hepatitis: Steatohepatitis and Viral Hepatitis
6. Liver Fibrosis
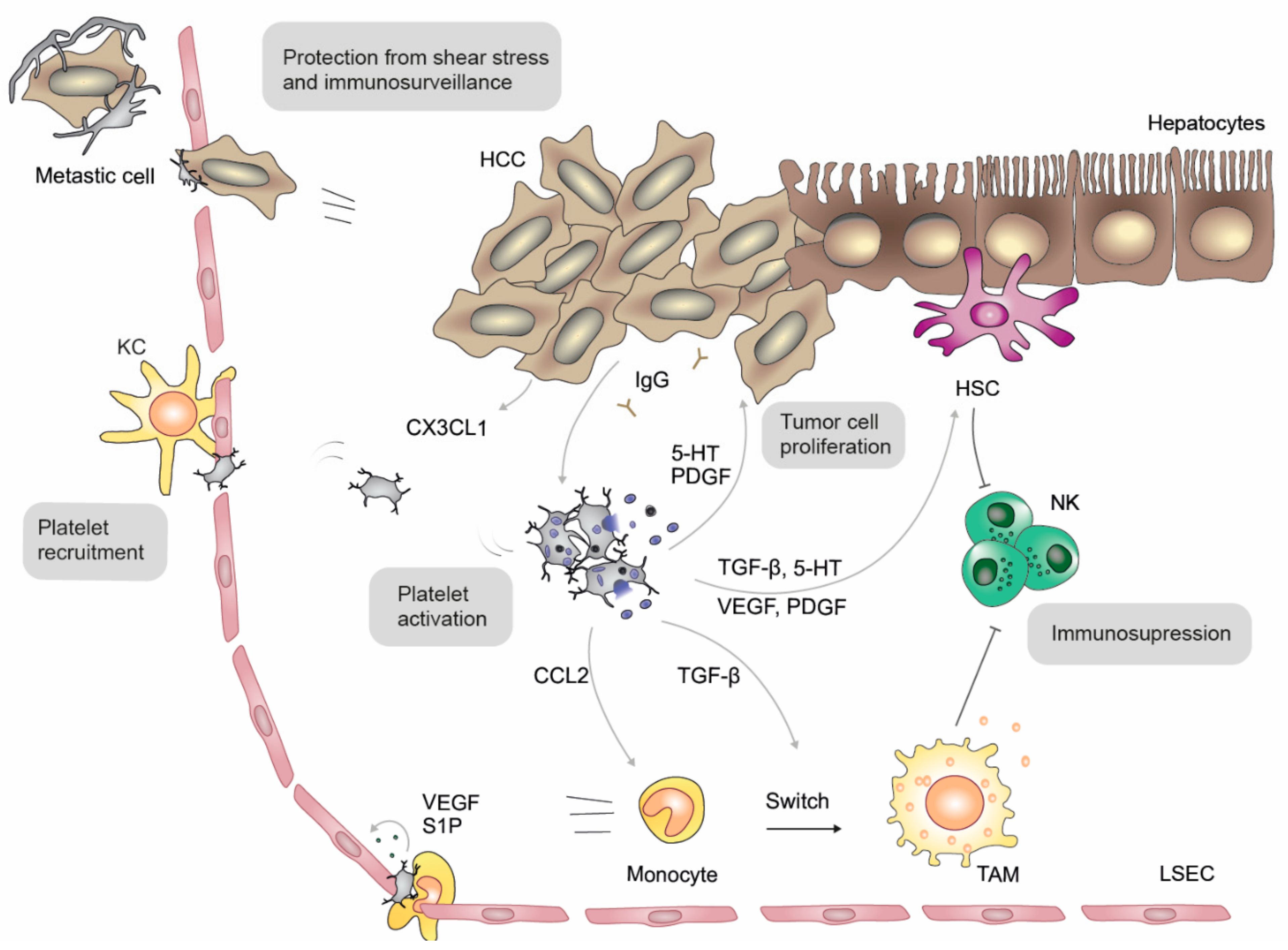
7. Hepatocellular Carcinoma (HCC)
8. Liver Regeneration
9. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- De Sauvage, F.J.; Hass, P.E.; Spencer, S.D.; Malloy, B.E.; Gurney, A.L.; Spencer, S.A.; Darbonne, W.C.; Henzel, W.J.; Wong, S.C.; Kuang, W.-J.; et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nat. Cell Biol. 1994, 369, 533–538. [Google Scholar] [CrossRef]
- Gangireddy, V.; Kanneganti, P.; Sridhar, S.; Talla, S.; Coleman, T. Management of Thrombocytopenia in Advanced Liver Disease. Can. J. Gastroenterol. Hepatol. 2014, 28, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Bashour, F.N.; Teran, C.J.; Mullen, K.D. Prevalence of Peripheral Blood Cytopenias (Hypersplenism) in Patients with Nonalcoholic Chronic Liver Disease. Am. J. Gastroenterol. 2000, 95, 2936–2939. [Google Scholar] [CrossRef]
- Gotlieb, N.; Schwartz, N.; Zelber-Sagi, S.; Chodick, G.; Shalev, V.; Shibolet, O. Longitudinal decrease in platelet counts as a surrogate marker of liver fibrosis. World J. Gastroenterol. 2020, 26, 5849–5862. [Google Scholar] [CrossRef]
- Alkozai, E.M.; Nijsten, M.W.; de Jong, K.P.; de Boer, M.T.; Peeters, P.M.J.G.; Slooff, M.J.; Porte, R.J.; Lisman, T. Immediate Postoperative Low Platelet Count is Associated with Delayed Liver Function Recovery After Partial Liver Resection. Ann. Surg. 2010, 251, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Q.; Yang, J.; Yang, J.-Y.; Wang, W.-T.; Yan, L.-N. Low immediate postoperative platelet count is associated with hepatic insufficiency after hepatectomy. World J. Gastroenterol. 2014, 20, 11871–11877. [Google Scholar] [CrossRef]
- Damm, G.; Pfeiffer, E.; Burkhardt, B.; Vermehren, J.; Nussler, A.K.; Weiss, T.S. Human parenchymal and non-parenchymal liver cell isolation, culture and characterization. Hepatol. Int. 2013, 7, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Starlinger, P.; Haegele, S.; Offensperger, F.; Oehlberger, L.; Pereyra, D.; Kral, J.B.; Schrottmaier, W.C.; Badrnya, S.; Reiberger, T.; Ferlitsch, A.; et al. The profile of platelet α-granule released molecules affects postoperative liver regeneration. Hepatology 2016, 63, 1675–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Støy, S.; Patel, V.C.; Sturgeon, J.P.; Vijay, G.K.M.; Lisman, T.; Bernal, W.; Shawcross, D.L. Platelet-leucocyte aggregation is augmented in cirrhosis and further increased by platelet transfusion. Aliment. Pharmacol. Ther. 2018, 47, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed Anuclear Cell Death Delimits Platelet Life Span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Palomo, I.; Ocqueteau, M.; Soto, M.; Aranda, E.; Mezzano, D.; Pereira, J. Platelet Aging In Vivo Is Associated with Loss of Membrane Phospholipid Asymmetry. Thromb. Haemost. 1999, 82, 1318–1321. [Google Scholar] [CrossRef]
- Sørensen, A.L.; Rumjantseva, V.; Nayeb-Hashemi, S.; Clausen, H.; Hartwig, J.H.; Wandall, H.H.; Hoffmeister, K.M. Role of sialic acid for platelet life span: Exposure of β-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor–expressing liver macrophages and hepatocytes. Blood 2009, 114, 1645–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boza, C.; Riquelme, A.; Ibañez, L.; Duarte, I.; Norero, E.; Viviani, P.; Soza, A.; Fernandez, J.I.; Raddatz, A.; Guzman, S.; et al. Predictors of Nonalcoholic Steatohepatitis (NASH) in Obese Patients Undergoing Gastric Bypass. Obes. Surg. 2005, 15, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.-Y.; Park, E.-J.; Lee, C.-W. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp. Mol. Med. 2020, 52, 1209–1219. [Google Scholar] [CrossRef]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic Fatty Liver Disease in Lean Individuals in the United States. Medicine (Baltimore) 2012, 91, 319–327. [Google Scholar] [CrossRef]
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordeaux, B.C.; Qayyum, R.; Yanek, L.R.; Vaidya, D.; Becker, L.C.; Faraday, N.; Becker, D.M. Effect of Obesity on Platelet Reactivity and Response to Low-Dose Aspirin. Prev. Cardiol. 2010, 13, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.N.; Sueiro, A.M.; Izquierdo, I.; Hermida-Nogueira, L.; Guitián, E.; Casanueva, F.F.; Farndale, R.W.; Moroi, M.; Jung, S.M.; Pardo, M.; et al. GPVI surface expression and signalling pathway activation are increased in platelets from obese patients: Elucidating potential anti-atherothrombotic targets in obesity. Atherosclerosis 2019, 281, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamminen, M.; Lassila, R.; Westerbacka, J.; Vehkavaara, S.; Yki-Järvinen, H. Obesity is associated with impaired platelet-inhibitory effect of acetylsalicylic acid in nondiabetic subjects. Int. J. Obes. 2003, 27, 907–911. [Google Scholar] [CrossRef] [Green Version]
- Bodary, P.F.; Westrick, R.J.; Wickenheiser, K.J.; Shen, Y.; Eitzman, D.T. Effect of leptin on arterial thrombosis following vascular injury in mice. JAMA 2002, 287, 1706–1709. [Google Scholar] [CrossRef] [Green Version]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.-S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsonello, A.; Perticone, F.; Malara, A.; De Domenico, D.; Loddo, S.; Buemi, M.; Ientile, R.; Corica, F. Leptin-dependent platelet aggregation in healthy, overweight and obese subjects. Int. J. Obes. 2003, 27, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Corsonello, A.; Malara, A.; Ientile, R.; Corica, F. Leptin enhances adenosine diphosphate-induced platelet aggregation in healthy subjects. Obes. Res. 2002, 10, 306. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chen, J.; Mao, J.; Li, H.; Wang, M.; Zhang, H.; Li, H.; Chen, W. Genistein Ameliorates Non-alcoholic Fatty Liver Disease by Targeting the Thromboxane A2 Pathway. J. Agric. Food Chem. 2018, 66, 5853–5859. [Google Scholar] [CrossRef]
- Roberts, L.N.; Bernal, W. Incidence of Bleeding and Thrombosis in Patients with Liver Disease. Semin. Thromb. Hemost. 2020, 46, 656–664. [Google Scholar] [CrossRef]
- Di Minno, M.N.; Tufano, A.; Rusolillo, A.; Di Minno, G.; Tarantino, G. High prevalence of nonalcoholic fatty liver in patients with idiopathic venous thromboembolism. World J. Gastroenterol. 2010, 16, 6119–6122. [Google Scholar] [CrossRef]
- Stine, J.G.; Shah, N.L.; Argo, C.K.; Pelletier, S.J.; Caldwell, S.H.; Northup, P.G. Increased risk of portal vein thrombosis in patients with cirrhosis due to nonalcoholic steatohepatitis. Liver Transplant. 2015, 21, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Potze, W.; Siddiqui, M.S.; Boyett, S.L.; Adelmeijer, J.; Daita, K.; Sanyal, A.J.; Lisman, T. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 65, 980–987. [Google Scholar] [CrossRef]
- Dunn, E.J.; Philippou, H.; Ariëns, R.A.S.; Grant, P.J. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetology 2006, 49, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, T.; Jenne, C.N. Fibrin fuels fatty liver disease. J. Thromb. Haemost. 2018, 16, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopec, A.K.; Joshi, N.; Towery, K.L.; Kassel, K.M.; Sullivan, B.P.; Flick, M.J.; Luyendyk, J.P. Thrombin Inhibition with Dabigatran Protects against High-Fat Diet–Induced Fatty Liver Disease in Mice. J. Pharmacol. Exp. Ther. 2014, 351, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kassel, K.M.; Owens, A.P.; Rockwell, C.E.; Sullivan, B.P.; Wang, R.; Tawfik, O.; Li, G.; Guo, G.L.; Mackman, N.; Luyendyk, J.P. Protease-Activated Receptor 1 and Hematopoietic Cell Tissue Factor Are Required for Hepatic Steatosis in Mice Fed a Western Diet. Am. J. Pathol. 2011, 179, 2278–2289. [Google Scholar] [CrossRef]
- Bai, J.; Xia, M.; Xue, Y.; Ma, F.; Cui, A.; Sun, Y.; Han, Y.; Xu, X.; Zhang, F.; Hu, Z.; et al. Thrombospondin 1 improves hepatic steatosis in diet-induced insulin-resistant mice and is associated with hepatic fat content in humans. EBioMedicine 2020, 57, 102849. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; Martino, J.S.; Pirola, C.J. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis 2010, 209, 585–591. [Google Scholar] [CrossRef]
- Taipale, T.; Seppälä, I.; Raitoharju, E.; Mononen, N.; Lyytikäinen, L.-P.; Illig, T.; Waldenberger, M.; Juonala, M.; Hutri-Kähönen, N.; Oksala, N.; et al. Fatty liver is associated with blood pathways of inflammatory response, immune system activation and prothrombotic state in Young Finns Study. Sci. Rep. 2018, 8, 10358. [Google Scholar] [CrossRef]
- Poggi, M.; Engel, D.; Christ, A.; Beckers, L.; Wijnands, E.; Boon, L.; Driessen, A.; Cleutjens, J.; Weber, C.; Gerdes, N.; et al. CD40L Deficiency Ameliorates Adipose Tissue Inflammation and Metabolic Manifestations of Obesity in Mice. Arter. Thromb. Vasc. Biol. 2011, 31, 2251–2260. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, J.; Lepreux, S.; Mulot, A.; Bérard, A.M.; Higa-Nishiyama, A.; Costet, P.; De Lédinghen, V.; Bioulac-Sage, P.; Balabaud, C.; Nurden, A.T.; et al. A protective role for CD154 in hepatic steatosis in mice. Hepatology 2010, 52, 1968–1979. [Google Scholar] [CrossRef]
- Lasagni, L.; Francalanci, M.; Annunziato, F.; Lazzeri, E.; Giannini, S.; Cosmi, L.; Sagrinati, C.; Mazzinghi, B.; Orlando, C.; Maggi, E.; et al. An Alternatively Spliced Variant of CXCR3 Mediates the Inhibition of Endothelial Cell Growth Induced by IP-10, Mig, and I-TAC, and Acts as Functional Receptor for Platelet Factor 4. J. Exp. Med. 2003, 197, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, J.; Man, K.; Li, X.; Du, J.; Chu, E.S.; Go, M.Y.; Sung, J.J.; Yu, J. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J. Hepatol. 2016, 64, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starlinger, P.; Pereyra, D.; Hackl, H.; Ortmayr, G.; Braunwarth, E.; Santol, J.; Najarnia, S.; Driedger, M.R.; Gregory, L.; Alva-Ruiz, R.; et al. Consequences of Perioperative Serotonin Reuptake Inhibitor Treatment during Hepatic Surgery. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Namkung, J.; Hwang, I.; Kim, H.; Lim, A.; Park, H.J.; Lee, H.W.; Han, K.-H.; Park, S.; Jeong, J.-S.; et al. Serotonin signals through a gut-liver axis to regulate hepatic steatosis. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nat. Cell Biol. 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Raparelli, V.; Basili, S.; Carnevale, R.; Napoleone, L.; Del Ben, M.; Nocella, C.; Bartimoccia, S.; Lucidi, C.; Talerico, G.; Riggio, O.; et al. Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology 2017, 65, 571–581. [Google Scholar] [CrossRef]
- Schattner, M. Platelet toll-like receptors in thromboinflammation. Front. Biosci. 2017, 22, 1867–1883. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the Liver by Intravascular Effector CD8 + T Cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated with Reduced Risk For Fibrosis Progression In Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.; Das, S.; Yadav, G.; Chaudhary, S.; Vyas, A.; Islam, M.; Gupta, A.C.; Bajpai, M.; Maiwall, R.; Maras, J.S.; et al. Hyperoxidized Albumin Modulates Platelets and Promotes Inflammation Through CD36 Receptor in Severe Alcoholic Hepatitis. Hepatol. Commun. 2020, 4, 50–65. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, C.; Ji, J.; Wang, C.; Yang, J.; Geng, B.; Zhao, T.; Zhou, H.; Mu, X.; Pan, J.; et al. CD36 deficiency attenuates immune-mediated hepatitis in mice by modulating the proapoptotic effects of CXC chemokine ligand 10. Hepatology 2018, 67, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.A.; Contaldo, C.; Georgiev, P.; El-Badry, A.M.; Recher, M.; Kurrer, M.; Cervantes-Barragan, L.; Ludewig, B.; Calzascia, T.; Bolinger, B.; et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat. Med. 2008, 14, 756–761. [Google Scholar] [CrossRef]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.R.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets mediate cytotoxic T lymphocyte–induced liver damage. Nat. Med. 2005, 11, 1167–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannacone, M.; Sitia, G.; Narvaiza, I.; Ruggeri, Z.M.; Guidotti, L.G. Antiplatelet Drug Therapy Moderates Immune-Mediated Liver Disease and Inhibits Viral Clearance in Mice Infected with a Replication-Deficient Adenovirus. Clin. Vaccine Immunol. 2007, 14, 1532–1535. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.G.; Duberg, A.-S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N. Engl. J. Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils Recruited to Sites of Infection Protect from Virus Challenge by Releasing Neutrophil Extracellular Traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariede, J.R.; Pardini, M.I.D.M.C.; Silva, G.F.; Grotto, R.M.T. Platelets can be a biological compartment for the Hepatitis C Virus. Braz. J. Microbiol. 2015, 46, 627–629. [Google Scholar] [CrossRef] [Green Version]
- Assinger, A. Platelets and Infection—An Emerging Role of Platelets in Viral Infection. Front. Immunol. 2014, 5, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Kurokawa, T.; Ohkohchi, N. Platelets in liver disease, cancer and regeneration. World J. Gastroenterol. 2017, 23, 3228–3239. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; You, H.; Wu, X.; Zhou, J.; Ou, X.; Jia, J. Platelets’ increase is associated with improvement of liver fibrosis in entecavir-treated chronic hepatitis B patients with significant liver fibrosis. Hepatol. Int. 2018, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Murata, S.; Takahashi, K.; Tamura, T.; Nozaki, R.; Ikeda, N.; Fukunaga, K.; Oda, T.; Sasaki, R.; Ohkohchi, N. Platelet transfusion improves liver function in patients with chronic liver disease and cirrhosis. Tohoku J. Exp. Med. 2013, 229, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, N.; Murata, S.; Maruyama, T.; Tamura, T.; Nozaki, R.; Kawasaki, T.; Fukunaga, K.; Oda, T.; Sasaki, R.; Homma, M.; et al. Platelet-derived adenosine 5′-triphosphate suppresses activation of human hepatic stellate cell: In vitro study. Hepatol. Res. 2011, 42, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; Tadokoro, S.; et al. Thrombocytopenia Exacerbates Cholestasis-Induced Liver Fibrosis in Mice. Gastroenterology 2010, 138, 2487–2498.e7. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-H.; Matsumoto, K.; Bessho, K.; Nakamura, T. Growth Inhibition and Apoptosis in Liver Myofibroblasts Promoted by Hepatocyte Growth Factor Leads to Resolution from Liver Cirrhosis. Am. J. Pathol. 2005, 166, 1017–1028. [Google Scholar] [CrossRef] [Green Version]
- Salem, N.A.; Hamza, A.; Alnahdi, H.; Ayaz, N. Biochemical and Molecular Mechanisms of Platelet-Rich Plasma in Ameliorating Liver Fibrosis Induced by Dimethylnitrosurea. Cell. Physiol. Biochem. 2018, 47, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Murata, S.; Fukunaga, K.; Ohkohchi, N. Human platelets inhibit liver fibrosis in severe combined immunodeficiency mice. World J. Gastroenterol. 2013, 19, 5250–5260. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nat. Cell Biol. 1989, 342, 440–443. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, X.-M.; Wu, Y.; Li, Y.-M.; Zheng, Y.-W.; Ohkohchi, N. Prominent effect of platelet on improvement of liver cirrhosis. AME Case Rep. 2020, 4, 14. [Google Scholar] [CrossRef]
- Kurokawa, T.; Murata, S.; Ohkohchi, N. Stable Liver Function during Long-Term Administration of Eltrombopag, a Thrombopoietin Receptor Agonist, in Patients with Chronic Liver Disease. Tohoku J. Exp. Med. 2016, 240, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Saab, S.; Brown, R.S. Management of Thrombocytopenia in Patients with Chronic Liver Disease. Dig. Dis. Sci. 2019, 64, 2757–2768. [Google Scholar] [CrossRef]
- Watanabe, M.; Murata, S.; Hashimoto, I.; Nakano, Y.; Ikeda, O.; Aoyagi, Y.; Matsuo, R.; Fukunaga, K.; Yasue, H.; Ohkohchi, N. Platelets contribute to the reduction of liver fibrosis in mice. J. Gastroenterol. Hepatol. 2009, 24, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Hashimoto, I.; Nakano, Y.; Myronovych, A.; Watanabe, M.; Ohkohchi, N. Single Administration of Thrombopoietin Prevents Progression of Liver Fibrosis and Promotes Liver Regeneration After Partial Hepatectomy in Cirrhotic Rats. Ann. Surg. 2008, 248, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto, T.; Eguchi, G.; Beebe, D.C. Alpha-smooth muscle actin expression in cultured lens epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1122–1129. [Google Scholar]
- Poujol-Robert, A.; Boëlle, P.-Y.; Conti, F.; Durand, F.; Duvoux, C.; Wendum, D.; Paradis, V.; Mackiewicz, V.; Chazouillères, O.; Corpechot, C.; et al. Aspirin may reduce liver fibrosis progression: Evidence from a multicenter retrospective study of recurrent hepatitis C after liver transplantation. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 570–576. [Google Scholar] [CrossRef]
- Jiang, Z.G.; Feldbrügge, L.; Tapper, E.B.; Popov, Y.; Ghaziani, T.; Afdhal, N.; Robson, S.C.; Mukamal, K.J. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment. Pharmacol. Ther. 2016, 43, 734–743. [Google Scholar] [CrossRef]
- Iqbal, U.; Dennis, B.B.; Li, A.A.; Cholankeril, G.; Kim, D.; Khan, M.A.; Ahmed, A. Use of anti-platelet agents in the prevention of hepatic fibrosis in patients at risk for chronic liver disease: A systematic review and meta-analysis. Hepatol. Int. 2019, 13, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Hussein, O.; Khalil, A.; Luder, A.; Szvalb, S.; Paizi, M.; Spira, G. The Beneficial Effect of Aspirin and Enoxaparin on Fibrosis Progression and Regenerative Activity in a Rat Model of Cirrhosis. Dig. Dis. Sci. 2007, 52, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.-W.; Chung, J.; Sverdlov, D.Y.; Miyamoto, M.; Kim, Y.O.; Ogawa, S.; Arch, R.H.; et al. Extrahepatic Platelet-Derived Growth Factor-β, Delivered by Platelets, Promotes Activation of Hepatic Stellate Cells and Biliary Fibrosis in Mice. Gastroenterology 2014, 147, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Zaldivar, M.M.; Pauels, K.; Von Hundelshausen, P.; Berres, M.-L.; Schmitz, P.; Bornemann, J.; Kowalska, M.A.; Gassler, N.; Streetz, K.L.; Weiskirchen, R.; et al. CXC chemokine ligand 4 (Cxcl4) is a platelet-derived mediator of experimental liver fibrosis. Hepatology 2009, 51, 1345–1353. [Google Scholar] [CrossRef]
- Sullivan, B.P.; Wang, R.; Tawfik, O.; Luyendyk, J.P. Protective and Damaging Effects of Platelets in Acute Cholestatic Liver Injury Revealed by Depletion and Inhibition Strategies. Toxicol. Sci. 2009, 115, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Mahmoud, N.I.; Messiha, B.A.; Salehc, I.G.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Interruption of platelets and thrombin function as a new approach against liver fibrosis induced experimentally in rats. Life Sci. 2019, 231, 116522. [Google Scholar] [CrossRef] [PubMed]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnman, N.; Francoz, C.; Barbu, V.; Wendum, D.; Rey, C.; Hultcrantz, R.; Poupon, R.; Housset, C. The Myofibroblastic Conversion of Peribiliary Fibrogenic Cells Distinct from Hepatic Stellate Cells Is Stimulated by Platelet-Derived Growth Factor During Liver Fibrogenesis. Lab. Investig. 2003, 83, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Yatomi, Y.; Yanase, M.; Satoh, H.; Maekawa, H.; Ogata, I.; Ozaki, Y.; Takuwa, Y.; Mochida, S.; Fujiwara, K. Biological activities of novel lipid mediator sphingosine 1-phosphate in rat hepatic stellate cells. Am. J. Physiol. Liver Physiol. 2000, 279, G304–G310. [Google Scholar] [CrossRef] [Green Version]
- Borkham-Kamphorst, E.; Van Roeyen, C.R.; Ostendorf, T.; Floege, J.; Gressner, A.M.; Weiskirchen, R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 2007, 46, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Kopec, A.K.; Ray, J.L.; Cline-Fedewa, H.; Groeneveld, D.J.; Lisman, T.; Luyendyk, J.P. Von Willebrand factor deficiency reduces liver fibrosis in mice. Toxicol. Appl. Pharmacol. 2017, 328, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaya, H.; Kawaratani, H.; Tsuji, Y.; Nakanishi, K.; Saikawa, S.; Sato, S.; Sawada, Y.; Kaji, K.; Okura, Y.; Shimozato, N.; et al. von Willebrand factor is a useful biomarker for liver fibrosis and prediction of hepatocellular carcinoma development in patients with hepatitis B and C. United Eur. Gastroenterol. J. 2018, 6, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Ruddell, R.G.; Oakley, F.; Hussain, Z.; Yeung, I.; Bryan-Lluka, L.J.; Ramm, G.A.; Mann, D.A. A Role for Serotonin (5-HT) in Hepatic Stellate Cell Function and Liver Fibrosis. Am. J. Pathol. 2006, 169, 861–876. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.C.; Jun, D.W.; Kwon, Y.I.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Yoon, B.C.; Choi, H.S.; Kim, E.K. 5-HT2A receptor antagonists inhibit hepatic stellate cell activation and facilitate apoptosis. Liver Int. 2013, 33, 535–543. [Google Scholar] [CrossRef]
- Ebrahimkhani, M.R.; Oakley, F.; Murphy, L.B.; Mann, J.; Moles, A.; Perugorria, M.J.; Ellis, E.L.; Lakey, A.F.; Burt, A.D.; Douglass, A.; et al. Stimulating healthy tissue regeneration by targeting the 5-HT2B receptor in chronic liver disease. Nat. Med. 2011, 17, 1668–1673. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Houlihan, D.D.; Kavanagh, D.; Haldar, D.; Luu, N.; Owen, A.; Suresh, S.; Ni Than, N.; Reynolds, G.; Penny, J.; et al. Sphingosine-1-Phosphate Prevents Egress of Hematopoietic Stem Cells from Liver to Reduce Fibrosis. Gastroenterology 2017, 153, 233–248.e16. [Google Scholar] [CrossRef] [Green Version]
- Rohrbach, T.; Maceyka, M.; Spiegel, S. Sphingosine kinase and sphingosine-1-phosphate in liver pathobiology. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Melki, I.; Tessandier, N.; Zufferey, A.; Boilard, E. Platelet microvesicles in health and disease. Platelets 2017, 28, 214–221. [Google Scholar] [CrossRef]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, B.; Kirstein, M.; Popp, S.; Hucke, F.; Bota, S.; Rohr-Udilova, N.; Reiberger, T.; Müller, C.; Trauner, M.; Peck-Radosavljevic, M.; et al. Association of Platelet Count and Mean Platelet Volume with Overall Survival in Patients with Cirrhosis and Unresectable Hepatocellular Carcinoma. Liver Cancer 2019, 8, 203–217. [Google Scholar] [CrossRef]
- Carr, B.I.; Guerra, V. Hepatocellular Carcinoma Size: Platelets, γ-Glutamyl Transpeptidase, and Alkaline Phosphatase. Oncology 2013, 85, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, S.; Lu, M.; Liu, Y.; Shu, D.; Zhu, Y.; Song, W.; Ma, Y.; Ma, R.; Zhang, B.; Fang, C.; et al. Platelets are recruited to hepatocellular carcinoma tissues in a CX3CL1-CX3CR1 dependent manner and induce tumour cell apoptosis. Mol. Oncol. 2020, 14, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Shu, D.; Zhu, Y.; Lu, M.; Zhang, Q.; Pei, Y.; He, A.-D.; Ma, R.; Zhang, B.; Ming, Z.-Y. Cancer cell-derived immunoglobulin G activates platelets by binding to platelet FcγRIIa. Cell Death Dis. 2019, 10, 87. [Google Scholar] [CrossRef]
- Bihari, C.; Rastogi, A.; Shasthry, S.M.; Bajpai, M.; Bhadoria, A.S.; Rajesh, S.; Mukund, A.; Kumar, A.; Sarin, S.K. Platelets contribute to growth and metastasis in hepatocellular carcinoma. APMIS 2016, 124, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wei, B.; Zhou, W.; Yang, Y.; Li, B.; Guo, S.; Li, J.; Ye, J.; Li, J.; Zhang, Q.; et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget 2015, 6, 6584–6596. [Google Scholar] [CrossRef] [Green Version]
- Lavergne, M.; Janus-Bell, E.; Schaff, M.; Gachet, C.; Mangin, P.H. Platelet Integrins in Tumor Metastasis: Do They Represent a Therapeutic Target? Cancers 2017, 9, 133. [Google Scholar] [CrossRef]
- Zhang, R.; Guo, H.; Xu, J.; Li, B.; Liu, Y.-J.; Cheng, C.; Zhou, C.; Zhao, Y.; Liu, Y. Activated platelets inhibit hepatocellular carcinoma cell differentiation and promote tumor progression via platelet-tumor cell binding. Oncotarget 2016, 7, 60609–60622. [Google Scholar] [CrossRef]
- Tesfamariam, B. Involvement of platelets in tumor cell metastasis. Pharmacol. Ther. 2016, 157, 112–119. [Google Scholar] [CrossRef]
- He, A.-D.; Xie, W.; Song, W.; Ma, Y.-Y.; Liu, G.; Liang, M.-L.; Da, X.-W.; Yao, G.-Q.; Zhang, B.-X.; Gao, C.-J.; et al. Platelet releasates promote the proliferation of hepatocellular carcinoma cells by suppressing the expression of KLF6. Sci. Rep. 2017, 7, 3989. [Google Scholar] [CrossRef] [Green Version]
- Soll, C.; Jang, J.H.; Riener, M.-O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.-A. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 2009, 51, 1244–1254. [Google Scholar] [CrossRef]
- Liu, S.; Miao, R.; Zhai, M.; Pang, Q.; Deng, Y.; Liu, S.; Qu, K.; Liu, C.; Zhang, J. Effects and related mechanisms of serotonin on malignant biological behavior of hepatocellular carcinoma via regulation of Yap. Oncotarget 2017, 8, 47412–47424. [Google Scholar] [CrossRef] [Green Version]
- Padickakudy, R.; Pereyra, D.; Offensperger, F.; Jonas, P.; Oehlberger, L.; Schwarz, C.; Haegele, S.; Assinger, A.; Brostjan, C.; Gruenberger, T.; et al. Bivalent role of intra-platelet serotonin in liver regeneration and tumor recurrence in humans. J. Hepatol. 2017, 67, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Nouso, K.; Wada, N.; Takeuchi, Y.; Kinugasa, H.; Miyahara, K.; Yasunaka, T.; Kuwaki, K.; Onishi, H.; Ikeda, F.; et al. Involvement of platelets in extrahepatic metastasis of hepatocellular carcinoma. Hepatol. Res. 2014, 44, E353–E359. [Google Scholar] [CrossRef] [Green Version]
- Carr, B.I.; Cavallini, A.; D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Mazzocca, A.; Messa, C. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Xin, G.; Wei, Z.; Li, S.; Xing, Z.; Ji, C.; Du, J.; Niu, H.; Huang, W. Dihydrodiosgenin inhibits endothelial cell-derived factor VIII and platelet-mediated hepatocellular carcinoma metastasis. Cancer Manag. Res. 2019, 11, 4871–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Murata, S.; Takahashi, K.; Nozaki, R.; Ohshiro, Y.; Ikeda, N.; Pak, S.; Myronovych, A.; Hisakura, K.; Fukunaga, K.; et al. Activation of human liver sinusoidal endothelial cell by human platelets induces hepatocyte proliferation. J. Hepatol. 2010, 53, 648–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokomori, H.; Oda, M.; Yoshimura, K.; Nagai, T.; Ogi, M.; Nomura, M.; Ishii, H. Vascular endothelial growth factor increases fenestral permeability in hepatic sinusoidal endothelial cells. Liver Int. 2003, 23, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.E.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, T.T.S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Zernecke, A.; Liehn, E.A.; Von Hundelshausen, P.; Knarren, S.; Kuziel, W.A.; Weber, C. Crucial Role of the CCL2/CCR2 Axis in Neointimal Hyperplasia After Arterial Injury in Hyperlipidemic Mice Involves Early Monocyte Recruitment and CCL2 Presentation on Platelets. Circ. Res. 2004, 95, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Vasina, E.M.; Cauwenberghs, S.; Feijge, M.A.H.; Heemskerk, J.W.M.; Weber, C.; Koenen, R.R. Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death Dis. 2011, 2, e211. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Ohkohchi, N.; Matsuo, R.; Ikeda, O.; Myronovych, A.; Hoshi, R. Platelets Promote Liver Regeneration in Early Period after Hepatectomy in Mice. World J. Surg. 2007, 31, 808–816. [Google Scholar] [CrossRef]
- Murata, S.; Matsuo, R.; Ikeda, O.; Myronovych, A.; Watanabe, M.; Hisakura, K.; Nakano, Y.; Hashimoto, I.; Ohkohchi, N. Platelets Promote Liver Regeneration under Conditions of Kupffer Cell Depletion after Hepatectomy in Mice. World J. Surg. 2008, 32, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Jenne, C.N.; Veldhuis, Z.J.; Sjollema, K.A.; Lenting, P.J.; Giepmans, B.N.G.; Porte, R.J.; Kubes, P.; Denis, C.V.; Lisman, T. Transient von Willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice. Liver Int. 2017, 37, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Storrie, B. The cellular basis of platelet secretion: Emerging structure/function relationships. Platelets 2017, 28, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Abu Rmilah, A.; Zhou, W.; Nelson, E.; Lin, L.; Amiot, B.; Nyberg, S.L. Understanding the marvels behind liver regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e340. [Google Scholar] [CrossRef]
- Gustafson, E.; Hamad, O.A.; Deckmyn, H.; Barbu, A.; Ekdahl, K.N.; Nilsson, B. Exposure of von Willebrand Factor on Isolated Hepatocytes Promotes Tethering of Platelets to the Cell Surface. Transplantation 2019, 103, 1630–1638. [Google Scholar] [CrossRef]
- Starlinger, P.; Pereyra, D.; Haegele, S.; Braeuer, P.; Oehlberger, L.; Primavesi, F.; Kohler, A.; Offensperger, F.; Reiberger, T.; Ferlitsch, A.; et al. Perioperative von Willebrand factor dynamics are associated with liver regeneration and predict outcome after liver resection. Hepatology 2018, 67, 1516–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, R.; Ohkohchi, N.; Murata, S.; Ikeda, O.; Nakano, Y.; Watanabe, M.; Hisakura, K.; Myronovych, A.; Kubota, T.; Narimatsu, H.; et al. Platelets Strongly Induce Hepatocyte Proliferation with IGF-1 and HGF In Vitro. J. Surg. Res. 2008, 145, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oe, H.; Kaido, T.; Mori, A.; Onodera, H.; Imamura, M. Hepatocyte growth factor as well as vascular endothelial growth factor gene induction effectively promotes liver regeneration after hepatectomy in Solt-Farber rats. Hepatogastroenterology 2005, 52, 1393–1397. [Google Scholar] [PubMed]
- Bockhorn, M.; Goralski, M.; Prokofiev, D.; Dammann, P.; Grünewald, P.; Trippler, M.; Biglarnia, A.; Kamler, M.; Niehues, E.M.; Frilling, A.; et al. VEGF is Important for Early Liver Regeneration After Partial Hepatectomy. J. Surg. Res. 2007, 138, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, D.; Pereyra, D.; Veldhuis, Z.; Adelmeijer, J.; Ottens, P.; Kopec, A.K.; Starlinger, P.; Lisman, T.; Luyendyk, J.P. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood 2019, 133, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Margonis, G.A.; Amini, N.; Buettner, S.; Besharati, S.; Kim, Y.; Sobhani, F.; Kamel, I.R.; Pawlik, T.M. Impact of early postoperative platelet count on volumetric liver gain and perioperative outcomes after major liver resection. BJS 2016, 103, 899–907. [Google Scholar] [CrossRef]
- Haegele, S.; Offensperger, F.; Pereyra, D.; Lahner, E.; Assinger, A.; Fleischmann, E.; Gruenberger, B.; Gruenberger, T.; Brostjan, C.; Starlinger, P. Deficiency in Thrombopoietin Induction after Liver Surgery Is Associated with Postoperative Liver Dysfunction. PLoS ONE 2015, 10, e0116985. [Google Scholar] [CrossRef] [Green Version]
- Abshagen, K.; Mertens, F.; Eipel, C.; Vollmar, B. Limited therapeutic efficacy of thrombopoietin on the regeneration of steatotic livers. Int. J. Clin. Exp. Pathol. 2013, 6, 1759–1769. [Google Scholar]
- Matsuo, R.; Nakano, Y.; Ohkohchi, N. Platelet Administration Via the Portal Vein Promotes Liver Regeneration in Rats After 70% Hepatectomy. Ann. Surg. 2011, 253, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, O.; Pehlivanli, F.; Karaca, G.; Aydin, G.; Altunkaya, C.; Bulut, H.; Aydın, O.; Pehlivanlı, F.; Aydın, G.; Aydın, O.; et al. May dexpanthenol, platelet-rich plasma, and thymoquinone provide new hope to maintain liver regeneration after partial hepatectomy? Turk. J. Gastroenterol. 2019, 30, 826–834. [Google Scholar] [CrossRef]
- López, M.L.; Kieling, C.O.; Cruz, C.U.; Osvaldt, A.; De Muñoz, G.O.; Meurer, L.; Silla, L.; Matte, U. Platelet increases survival in a model of 90% hepatectomy in rats. Liver Int. 2013, 34, 1049–1056. [Google Scholar] [CrossRef]
- Shimabukuro, R.; Kawanaka, H.; Tomikawa, M.; Akahoshi, T.; Konishi, K.; Yoshida, D.; Anegawa, G.; Uehara, H.; Hashimoto, N.; Hashizume, M.; et al. Effect of thrombopoietin on platelet counts and liver regeneration after partial hepatectomy in a rat model. Surg. Today 2009, 39, 1054–1059. [Google Scholar] [CrossRef]
- Myronovych, A.; Murata, S.; Chiba, M.; Matsuo, R.; Ikeda, O.; Watanabe, M.; Hisakura, K.; Nakano, Y.; Kohno, K.; Kawasaki, T.; et al. Role of platelets on liver regeneration after 90% hepatectomy in mice. J. Hepatol. 2008, 49, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.; Balaphas, A.; Fontana, P.; Morel, P.; Robson, S.C.; Sadoul, K.; Gonelle-Gispert, C.; Bühler, L. Platelet Interactions with Liver Sinusoidal Endothelial Cells and Hepatic Stellate Cells Lead to Hepatocyte Proliferation. Cells 2020, 9, 1243. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kozuma, Y.; Suzuki, H.; Tamura, T.; Maruyama, T.; Fukunaga, K.; Murata, S.; Ohkohchi, N. Human platelets promote liver regeneration with Kupffer cells in SCID mice. J. Surg. Res. 2013, 180, 62–72. [Google Scholar] [CrossRef]
- Tan, Q.; Hu, J.; Yu, X.; Guan, W.; Lu, H.; Yu, Y.; Yu, Y.; Zang, G.; Tang, Z. The Role of IL-1 Family Members and Kupffer Cells in Liver Regeneration. BioMed Res. Int. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shido, K.; Chavez, D.; Cao, Z.; Koji, S.; Rafii, S.; Ding, B.-S. Platelets prime hematopoietic–vascular niche to drive angiocrine-mediated liver regeneration. Signal Transduct. Target. Ther. 2017, 2, 16044. [Google Scholar] [CrossRef]
- Hoffmeister, K.M.; Felbinger, T.W.; Falet, H.; Denis, C.V.; Bergmeier, W.; Mayadas, T.N.; Von Andrian, U.H.; Wagner, D.D.; Stossel, T.P.; Hartwig, J.H. The Clearance Mechanism of Chilled Blood Platelets. Cell 2003, 112, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Kirschbaum, M.; Karimian, G.; Adelmeijer, J.; Giepmans, B.N.G.; Porte, R.J.; Lisman, T. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood 2015, 126, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.-A. Platelet-Derived Serotonin Mediates Liver Regeneration. Science 2006, 312, 104–107. [Google Scholar] [CrossRef]
- Sulaiman, P.; Joseph, B.; Kaimal, S.B.; Paulose, C.S. Decreased Hepatic 5-HT1A Receptors During Liver Regeneration and Neoplasia in Rats. Neurochem. Res. 2007, 33, 444–449. [Google Scholar] [CrossRef]
- Papadimas, G.K.; Tzirogiannis, K.N.; Panoutsopoulos, G.I.; Demonakou, M.D.; Skaltsas, S.D.; Hereti, R.I.; Papadopoulou-Daifoti, Z.; Mykoniatis, M.G. Effect of serotonin receptor 2 blockage on liver regeneration after partial hepatectomy in the rat liver. Liver Int. 2006, 26, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Furrer, K.; Rickenbacher, A.; Tian, Y.; Jochum, W.; Bittermann, A.G.; Käch, A.; Humar, B.; Graf, R.; Moritz, W.; Clavien, P.-A. Serotonin reverts age-related capillarization and failure of regeneration in the liver through a VEGF-dependent pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 2945–2950. [Google Scholar] [CrossRef] [Green Version]
- Starlinger, P.; Assinger, A.; Haegele, S.; Wanek, D.; Zikeli, S.; Schauer, D.; Birner, P.; Fleischmann, E.; Gruenberger, B.; Brostjan, C.; et al. Evidence for serotonin as a relevant inducer of liver regeneration after liver resection in humans. Hepatology 2014, 60, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Alkozai, E.M.; Van Faassen, M.; Kema, I.P.; Porte, R.J.; Lisman, T. Evidence against a role of serotonin in liver regeneration in humans. Hepatology 2015, 62, 983. [Google Scholar] [CrossRef] [Green Version]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105, S13–S33. [Google Scholar] [CrossRef]
- Peltekian, K.M.; Makowka, L.; Williams, R.; Blendis, L.M.; Levy, G.A. Prostaglandins in Liver Transplantation Research Group Prostaglandins in liver failure and transplantation: Regeneration, immunomodulation, and cytoprotection. Liver Transplant. Surg. 1996, 2, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Kopetz, S.; Hawk, E.; Sood, A.K.; Loree, J.M.; Gresele, P.; Honn, K.V. Platelet “first responders” in wound response, cancer, and metastasis. Cancer Metastasis Rev. 2017, 36, 199–213. [Google Scholar] [CrossRef]
- Aryal, B.; Yamakuchi, M.; Shimizu, T.; Kadono, J.; Furoi, A.; Gejima, K.; Komokata, T.; Hashiguchi, T.; Imoto, Y. Deciphering Platelet Kinetics in Diagnostic and Prognostic Evaluation of Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Asghar, S.; Waqar, W.; Umar, M.; Manzoor, S. Tumor educated platelets, a promising source for early detection of hepatocellular carcinoma: Liquid biopsy an alternative approach to tissue biopsy. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; Kooi, I.E.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhu, J.; Ma, X.; Wang, H.; Qiu, S.; Pan, B.; Zhou, J.; Fan, J.; Yang, X.; Guo, W.; et al. Platelet activation status in the diagnosis and postoperative prognosis of hepatocellular carcinoma. Clin. Chim. Acta 2019, 495, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hu, Q.; Jiang, C.; Gu, Z. Platelet for drug delivery. Curr. Opin. Biotechnol. 2019, 58, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zuo, H.; Chen, B.; Wang, R.; Ahmed, A.; Hu, Y.; Ouyang, J. Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7, 42632. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mussbacher, M.; Brunnthaler, L.; Panhuber, A.; Starlinger, P.; Assinger, A. Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 3113. https://doi.org/10.3390/ijms22063113
Mussbacher M, Brunnthaler L, Panhuber A, Starlinger P, Assinger A. Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases. International Journal of Molecular Sciences. 2021; 22(6):3113. https://doi.org/10.3390/ijms22063113
Chicago/Turabian StyleMussbacher, Marion, Laura Brunnthaler, Anja Panhuber, Patrick Starlinger, and Alice Assinger. 2021. "Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases" International Journal of Molecular Sciences 22, no. 6: 3113. https://doi.org/10.3390/ijms22063113
APA StyleMussbacher, M., Brunnthaler, L., Panhuber, A., Starlinger, P., & Assinger, A. (2021). Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases. International Journal of Molecular Sciences, 22(6), 3113. https://doi.org/10.3390/ijms22063113