
Members of the Fibroblast Growth Factor Receptor Superfamily Are Proteolytically Cleaved by Two Differently Activated Metalloproteases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
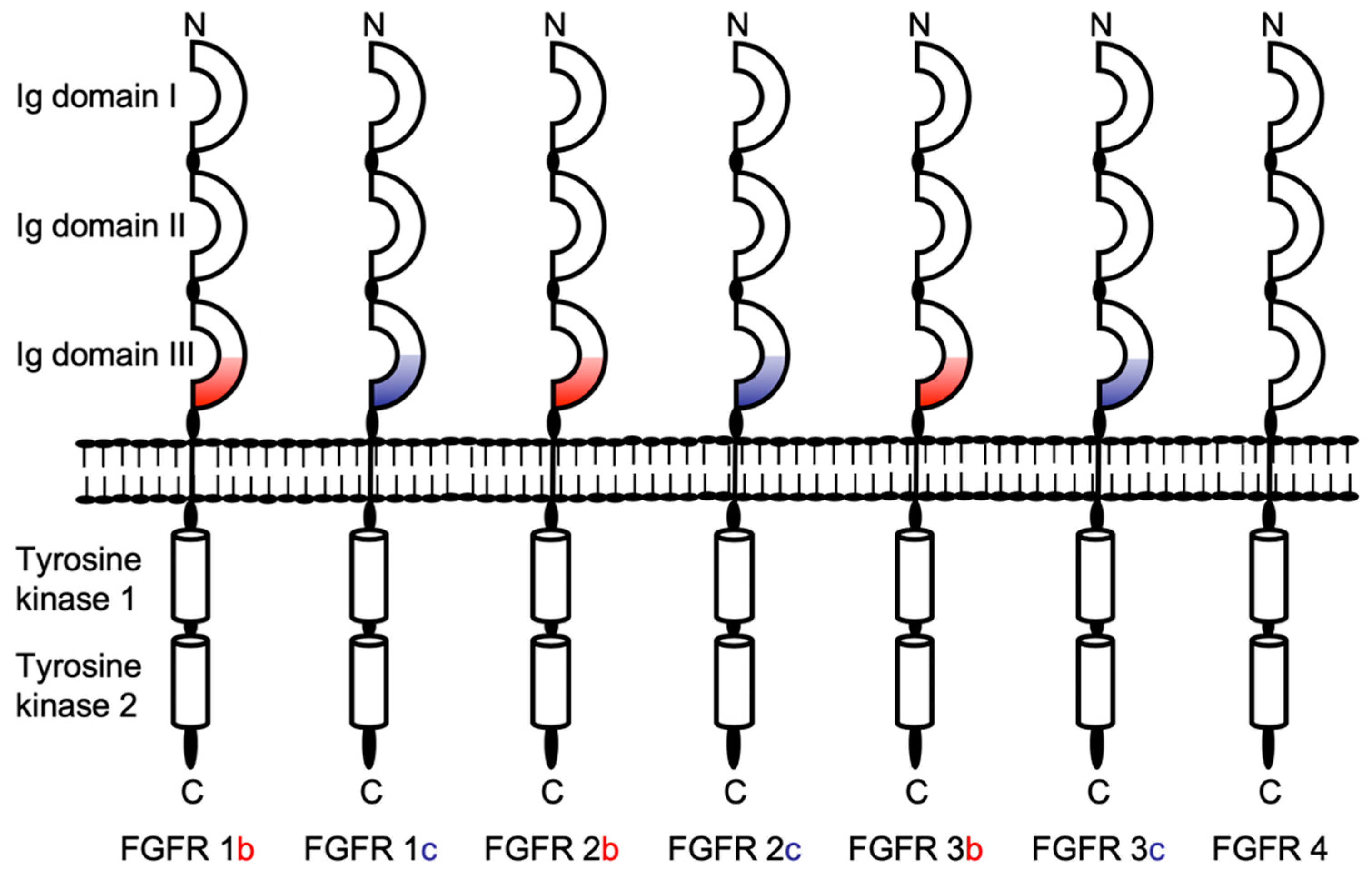
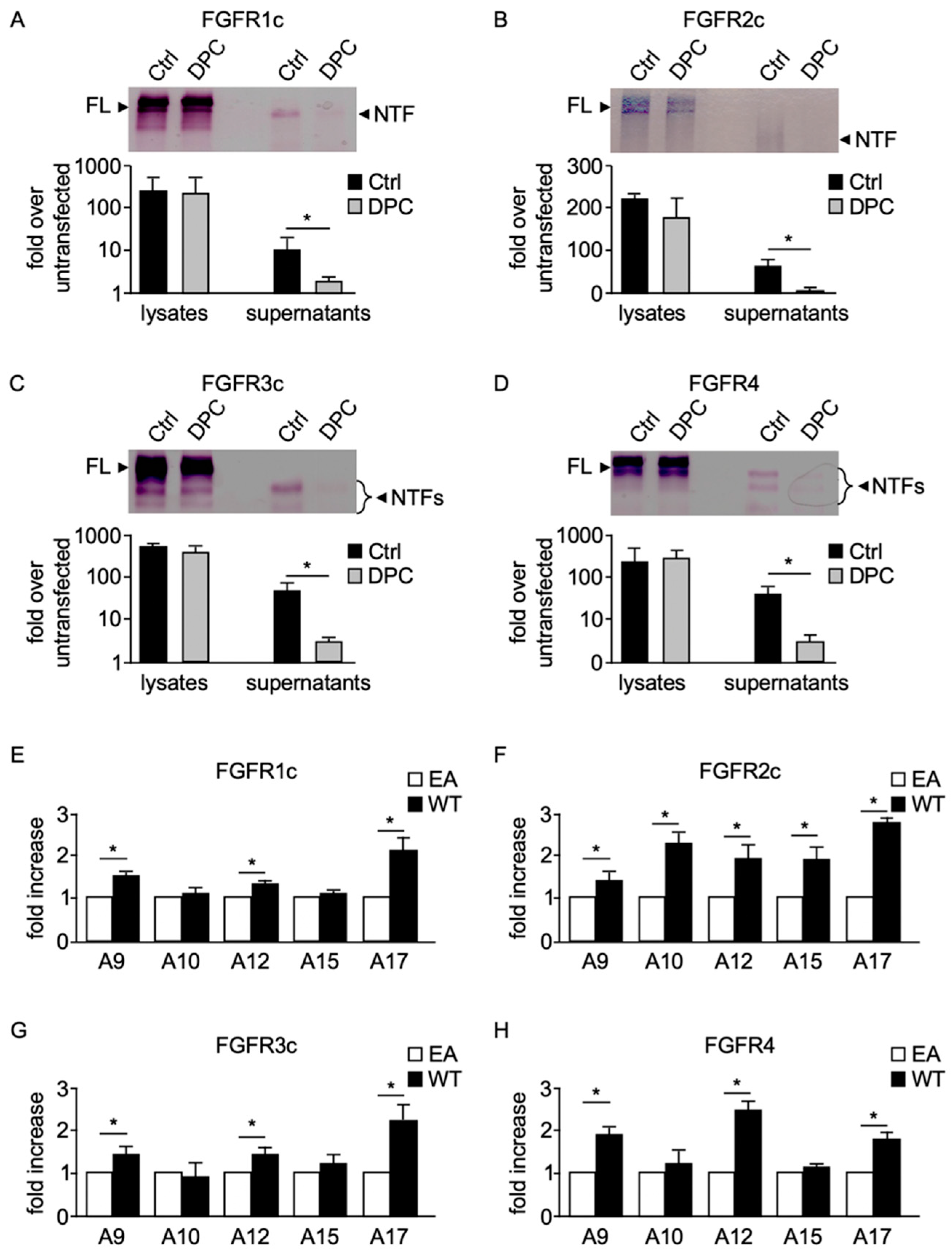
2.1. Ectodomains of Membrane-Anchored FGFRs Are Shed by a Metalloprotease Activity
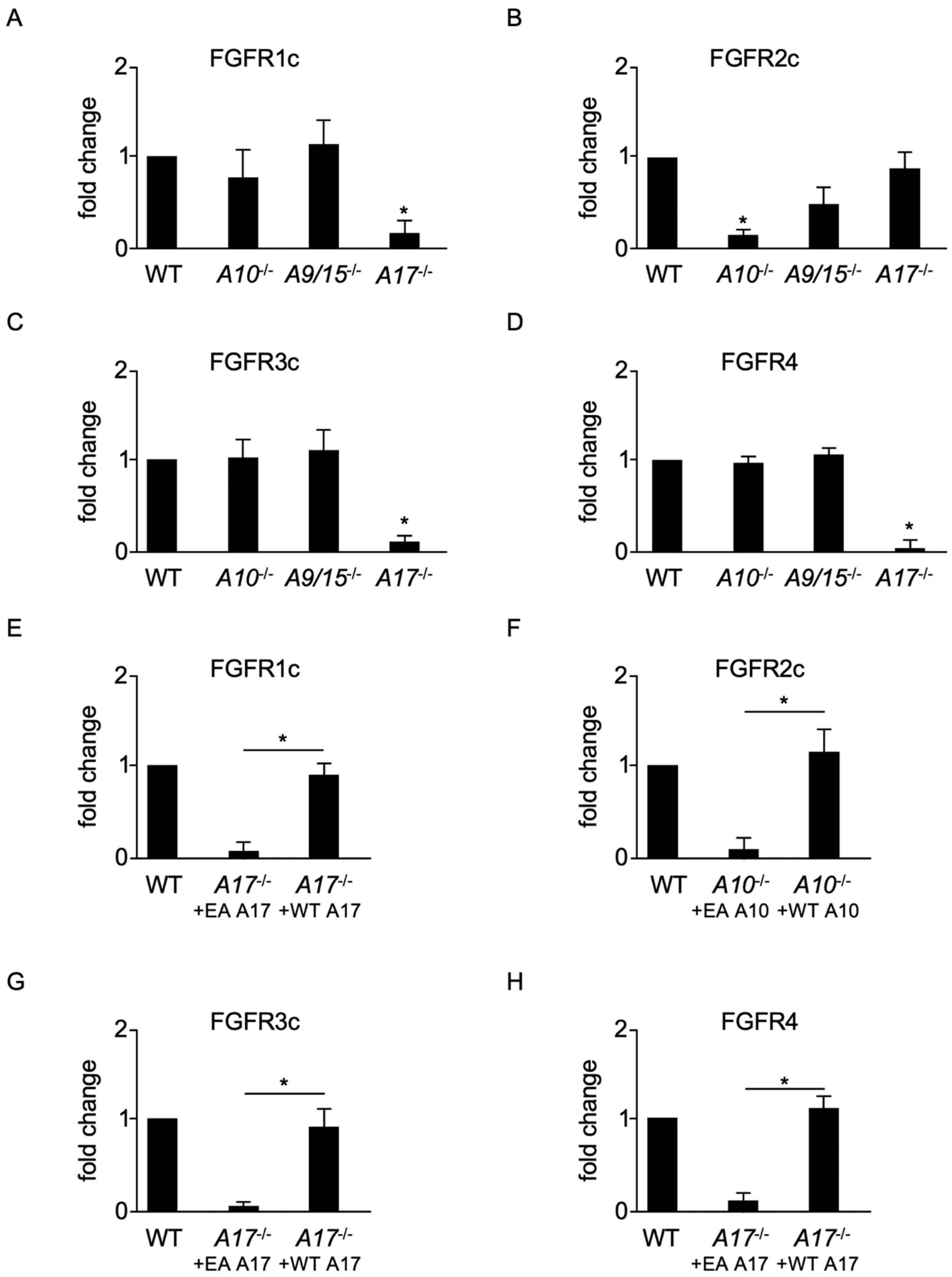
2.2. ADAMs 10 and 17 Are Involved in the Constitutive Shedding of Membrane-Anchored FGFRs
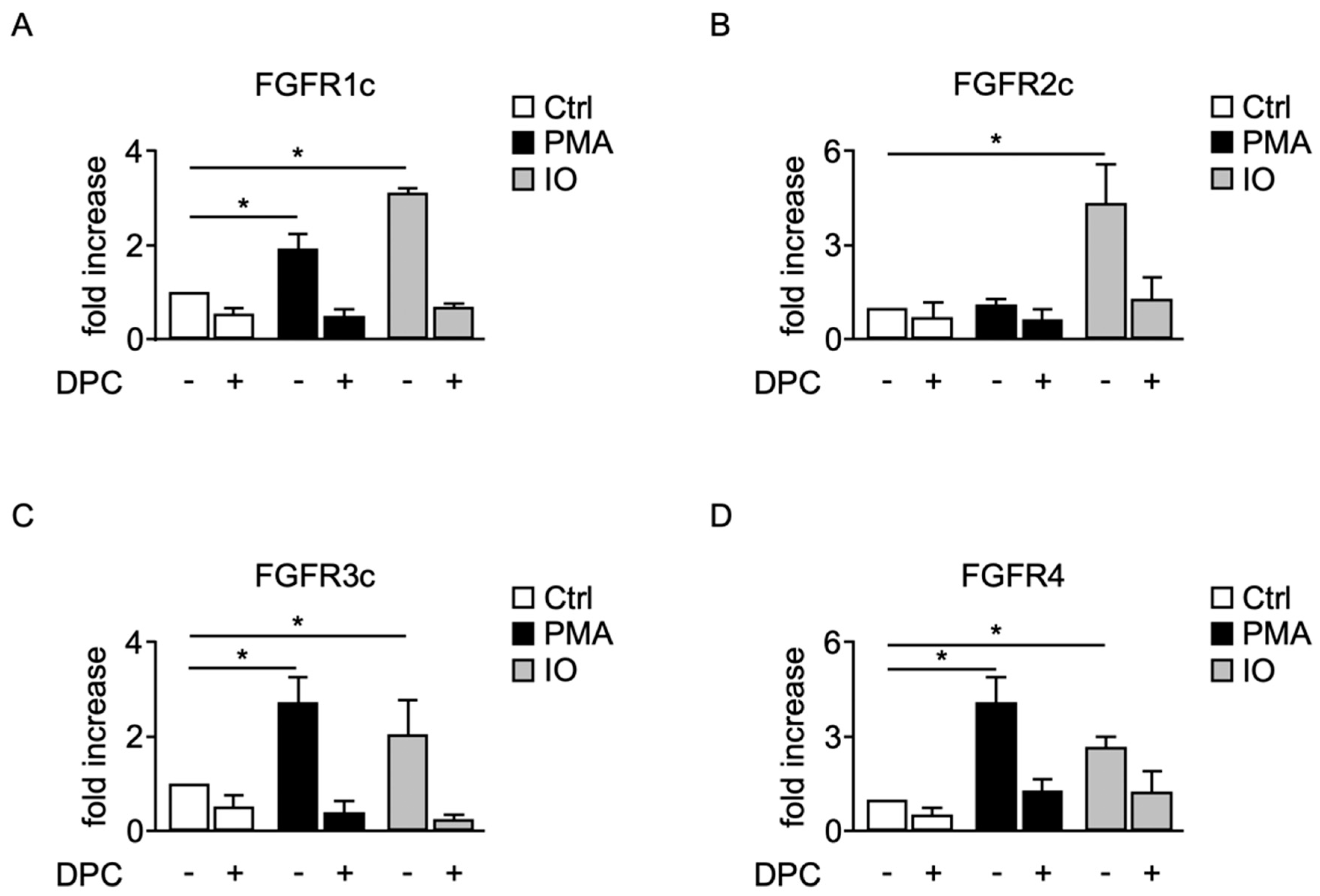
2.3. Shedding of Membrane-Anchored FGFRs Can Be Induced by the Protein Kinase C Activator PMA and the Calcium Ionophore Ionomycin
2.4. Induced Shedding of Membrane-Anchored FGFRs Depends on ADAMs 10 and 17
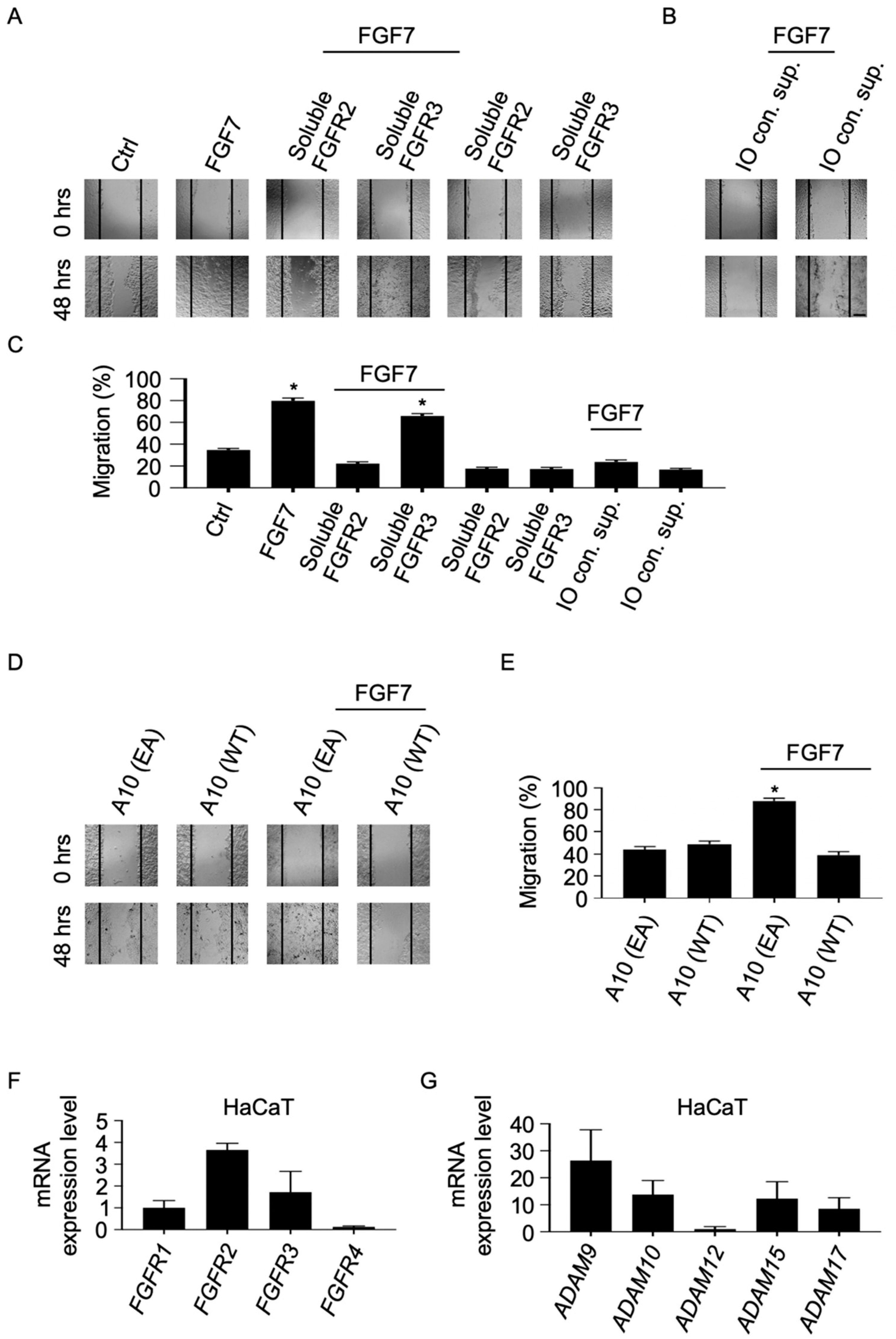
2.5. Soluble Forms of FGFR2 Inhibit FGF7-Induced Epithelial Cell Migration
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Expression Vectors
4.3. Transfection and Ectodomain Shedding Assays
4.4. In-Gel Alkaline Phosphatase Assay
4.5. In Vitro Scratch Wound-Healing Assays
4.6. Real-Time qRT-PCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM9 | A disintegrin and metalloprotease 9 |
| ADAM10 | A disintegrin and metalloprotease 10 |
| ADAM15 | A disintegrin and metalloprotease 15 |
| ADAM17 | A disintegrin and metalloprotease 17 |
| AP | Alkaline phosphatase |
| DPC333 | DPC333 ((2R)-2-((3R)-3amino-3(4-[2-methyl-4-quinolinyl)methoxyl] phenyl)-2-oxopyrrolidinyl)-N-hydroxy-4-methylpentanamide)) |
| EGFR | Epidermal growth factor receptor |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FGFR3 | Fibroblast growth factor receptor 3 |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FL | Full-length |
| IO | Ionomycin |
| mEF | Mouse embryonic fibroblast |
| NTF | N-terminal fragment |
| PMA | Phorbol 12-myristate 13-acetate |
| RTK | Receptor tyrosine kinase |
| WT | Wild type |
References
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.C.; Vaillancourt, R.R. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene 1998, 17, 1343–1352. [Google Scholar] [CrossRef] [Green Version]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreitman, M.; Noronha, A.; Yarden, Y. Irreversible modifications of receptor tyrosine kinases. FEBS Lett. 2018, 592, 2199–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [Green Version]
- Merilahti, J.A.M.; Elenius, K. Gamma-secretase-dependent signaling of receptor tyrosine kinases. Oncogene 2019, 38, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakabe, K.; Omura, T.; Shibagaki, Y.; Mihara, E.; Homma, K.; Kato, Y.; Yoshimura, A.; Murakami, Y.; Takagi, J.; Hattori, S.; et al. Mechanistic insights into ectodomain shedding: Susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci. Rep. 2017, 7, 46174. [Google Scholar] [CrossRef]
- Guillonneau, X.; Regnier-Ricard, F.; Laplace, O.; Jonet, L.; Bryckaert, M.; Courtois, Y.; Mascarelli, F. Fibroblast growth factor (FGF) soluble receptor 1 acts as a natural inhibitor of FGF2 neurotrophic activity during retinal degeneration. Mol. Biol. Cell 1998, 9, 2785–2802. [Google Scholar] [CrossRef] [Green Version]
- Chioni, A.M.; Grose, R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol. 2012, 197, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanneken, A.; Baird, A. Soluble forms of the high-affinity fibroblast growth factor receptor in human vitreous fluid. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1192–1196. [Google Scholar]
- Hanneken, A.; Maher, P.A.; Baird, A. High affinity immunoreactive FGF receptors in the extracellular matrix of vascular endothelial cells—Implications for the modulation of FGF-2. J. Cell Biol. 1995, 128, 1221–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanneken, A.; Ying, W.; Ling, N.; Baird, A. Identification of soluble forms of the fibroblast growth factor receptor in blood. Proc. Natl. Acad. Sci. USA 1994, 91, 9170–9174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanneken, A. Structural characterization of the circulating soluble FGF receptors reveals multiple isoforms generated by secretion and ectodomain shedding. FEBS Lett. 2001, 489, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Zhou, W.; Swendeman, S.L.; Wong, P.M.; Rafii, S.; Reiss, K.; Blobel, C.P. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun. 2011, 2, 229. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Reiss, K.; Maretzky, T.; Ludwig, A.; Tousseyn, T.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005, 24, 742–752. [Google Scholar] [CrossRef]
- Issuree, P.D.; Maretzky, T.; McIlwain, D.R.; Monette, S.; Qing, X.; Lang, P.A.; Swendeman, S.L.; Park-Min, K.H.; Binder, N.; Kalliolias, G.D.; et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Investig. 2013, 123, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Swendeman, S.; Mogollon, E.; Weskamp, G.; Sahin, U.; Reiss, K.; Blobel, C.P. Characterization of the catalytic properties of the membrane-anchored metalloproteinase ADAM9 in cell-based assays. Biochem. J. 2017, 474, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, N.; Horiuchi, K.; Becherer, J.D.; Toyama, Y.; Besmer, P.; Blobel, C.P. Different ADAMs have distinct influences on Kit ligand processing: Phorbol-ester-stimulated ectodomain shedding of Kitl1 by ADAM17 is reduced by ADAM19. J. Cell Sci. 2007, 120 Pt 6, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Prenzel, N.; Ullrich, A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol. Cell Biol. 2004, 24, 5172–5183. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Yang, G.; Ouerfelli, O.; Overall, C.M.; Worpenberg, S.; Hassiepen, U.; Eder, J.; Blobel, C.P. Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem. J. 2009, 420, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef] [Green Version]
- Groot, A.J.; Vooijs, M.A. The role of Adams in Notch signaling. Adv. Exp. Med. Biol. 2012, 727, 15–36. [Google Scholar] [PubMed] [Green Version]
- Briso, E.M.; Dienz, O.; Rincon, M. Cutting edge: Soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J. Immunol. 2008, 180, 7102–7106. [Google Scholar] [CrossRef] [Green Version]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Merilahti, J.A.M.; Ojala, V.K.; Knittle, A.M.; Pulliainen, A.T.; Elenius, K. Genome-wide screen of gamma-secretase-mediated intramembrane cleavage of receptor tyrosine kinases. Mol. Biol. Cell 2017, 28, 3123–3131. [Google Scholar] [CrossRef]
- Overall, C.M.; Blobel, C.P. In search of partners: Linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 2007, 8, 245–257. [Google Scholar] [CrossRef]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lubke, T.; Lena Illert, A.; von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [Green Version]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.; Niu, X.D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123 Pt 22, 3913–3922. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Morita, J.; Nakamura, M.; Kobayashi, Y.; Deng, C.X.; Funato, N.; Moriyama, K. Soluble form of FGFR2 with S252W partially prevents craniosynostosis of the apert mouse model. Dev. Dyn. 2014, 243, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, Y.; Yokozeki, M.; Hiura, K.; Matsumoto, K.; Nakanishi, H.; Matsumoto, T.; Marie, P.J.; Moriyama, K. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J. Biol. Chem. 2004, 279, 45926–45934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celli, G.; LaRochelle, W.J.; Mackem, S.; Sharp, R.; Merlino, G. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J. 1998, 17, 1642–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiong, K.H.; Mah, L.Y.; Leong, C.O. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis 2013, 18, 1447–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [Green Version]
- Komi-Kuramochi, A.; Kawano, M.; Oda, Y.; Asada, M.; Suzuki, M.; Oki, J.; Imamura, T. Expression of fibroblast growth factors and their receptors during full-thickness skin wound healing in young and aged mice. J. Endocrinol. 2005, 186, 273–289. [Google Scholar] [CrossRef]
- Qian, M.; Bai, S.A.; Brogdon, B.; Wu, J.T.; Liu, R.Q.; Covington, M.B.; Vaddi, K.; Newton, R.C.; Fossler, M.J.; Garner, C.E.; et al. Pharmacokinetics and pharmacodynamics of DPC 333 ((2R)-2-((3R)-3-amino-3{4-[2-methyl-4-quinolinyl) methoxy] phenyl}-2-oxopyrrolidinyl)-N-hydroxy-4-methylpentanamide)), a potent and selective inhibitor of tumor necrosis factor alpha-converting enzyme in rodents, dogs, chimpanzees, and humans. Drug Metab. Dispos. 2007, 35, 1916–1925. [Google Scholar]
- Maretzky, T.; Zhou, W.; Huang, X.Y.; Blobel, C.P. A transforming Src mutant increases the bioavailability of EGFR ligands via stimulation of the cell-surface metalloproteinase ADAM17. Oncogene 2011, 30, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt B, 2059–2070. [Google Scholar] [CrossRef]
- Chow, F.L.; Fernandez-Patron, C. Many membrane proteins undergo ectodomain shedding by proteolytic cleavage. Does one sheddase do the job on all of these proteins? IUBMB Life 2007, 59, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456. [Google Scholar] [CrossRef]
- Kato, T.; Hagiyama, M.; Ito, A. Renal ADAM10 and 17: Their Physiological and Medical Meanings. Front. Cell Dev. Biol. 2018, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Rustagi, S.; Dempsey, P.J. ADAM Proteases and Gastrointestinal Function. Annu. Rev. Physiol. 2016, 78, 243–276. [Google Scholar] [CrossRef] [Green Version]
- Garcia, S.; Dirat, B.; Tognacci, T.; Rochet, N.; Mouska, X.; Bonnafous, S.; Patouraux, S.; Tran, A.; Gual, P.; Le Marchand-Brustel, Y.; et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci. Transl. Med. 2013, 5, 203ra124. [Google Scholar] [CrossRef] [PubMed]
- Sturla, L.M.; Merrick, A.E.; Burchill, S.A. FGFR3IIIS: A novel soluble FGFR3 spliced variant that modulates growth is frequently expressed in tumour cells. Br. J. Cancer 2003, 89, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Heinzle, C.; Erdem, Z.; Paur, J.; Grasl-Kraupp, B.; Holzmann, K.; Grusch, M.; Berger, W.; Marian, B. Is fibroblast growth factor receptor 4 a suitable target of cancer therapy? Curr. Pharm. Des. 2014, 20, 2881–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, S.M.; Bobe, P.; Reiss, K.; Horiuchi, K.; Niu, X.D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Weskamp, G.; Cai, H.; Brodie, T.A.; Higashyama, S.; Manova, K.; Ludwig, T.; Blobel, C.P. Mice lacking the metalloprotease-disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol. Cell Biol. 2002, 22, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Weskamp, G.; Ford, J.W.; Sturgill, J.; Martin, S.; Docherty, A.J.; Swendeman, S.; Broadway, N.; Hartmann, D.; Saftig, P.; Umland, S.; et al. ADAM10 is a principal ‘sheddase’ of the low-affinity immunoglobulin E receptor CD23. Nat. Immunol. 2006, 7, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, C.; Klitgaard, M.; Noer, J.B.; Kotzsch, A.; Nehammer, C.; Kronqvist, P.; Berthelsen, J.; Blobel, C.; Kveiborg, M.; Albrechtsen, R.; et al. ADAM12 is expressed in the tumour vasculature and mediates ectodomain shedding of several membrane-anchored endothelial proteins. Biochem. J. 2013, 452, 97–109. [Google Scholar] [CrossRef]
- Chesneau, V.; Becherer, J.D.; Zheng, Y.; Erdjument-Bromage, H.; Tempst, P.; Blobel, C.P. Catalytic properties of ADAM19. J. Biol. Chem. 2003, 278, 22331–22340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Weskamp, G.; Zheng, Y.; Chesneau, V.; Horiuchi, K.; Blobel, C.P. A sensitive method to monitor ectodomain shedding of ligands of the epidermal growth factor receptor. Methods Mol. Biol. 2006, 327, 99–113. [Google Scholar]
- Zheng, Y.; Schlondorff, J.; Blobel, C.P. Evidence for regulation of the tumor necrosis factor alpha-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J. Biol. Chem. 2002, 277, 42463–42470. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Le Gall, S.; Alabi, R.O.; Speck, N.; Reiss, K.; Blobel, C.P. The cytoplasmic domain of a disintegrin and metalloproteinase 10 (ADAM10) regulates its constitutive activity but is dispensable for stimulated ADAM10-dependent shedding. J. Biol. Chem. 2015, 290, 7416–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixit, G.; Schanz, W.; Pappas, B.A.; Maretzky, T. Members of the Fibroblast Growth Factor Receptor Superfamily Are Proteolytically Cleaved by Two Differently Activated Metalloproteases. Int. J. Mol. Sci. 2021, 22, 3165. https://doi.org/10.3390/ijms22063165
Dixit G, Schanz W, Pappas BA, Maretzky T. Members of the Fibroblast Growth Factor Receptor Superfamily Are Proteolytically Cleaved by Two Differently Activated Metalloproteases. International Journal of Molecular Sciences. 2021; 22(6):3165. https://doi.org/10.3390/ijms22063165
Chicago/Turabian StyleDixit, Garima, Willow Schanz, Benjamin A. Pappas, and Thorsten Maretzky. 2021. "Members of the Fibroblast Growth Factor Receptor Superfamily Are Proteolytically Cleaved by Two Differently Activated Metalloproteases" International Journal of Molecular Sciences 22, no. 6: 3165. https://doi.org/10.3390/ijms22063165
APA StyleDixit, G., Schanz, W., Pappas, B. A., & Maretzky, T. (2021). Members of the Fibroblast Growth Factor Receptor Superfamily Are Proteolytically Cleaved by Two Differently Activated Metalloproteases. International Journal of Molecular Sciences, 22(6), 3165. https://doi.org/10.3390/ijms22063165