
Involvement of Cytokines in the Pathogenesis of Diabetic Macular Edema


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathogenesis
2.1. Biochemical Pathways
2.2. VEGF and VEGF Receptors
2.3. Inflammation
Effect of Inflammation on Blood Flow
3. Soluble Mediators Involved in DME
3.1. Growth Factors
3.1.1. PlGF
3.1.2. PDGF
3.2. Cytokines and Chemokines
3.2.1. IL-6
3.2.2. IL-8
3.2.3. MCP-1
3.2.4. IP-10
3.3. Other Mediators
ICAM-1
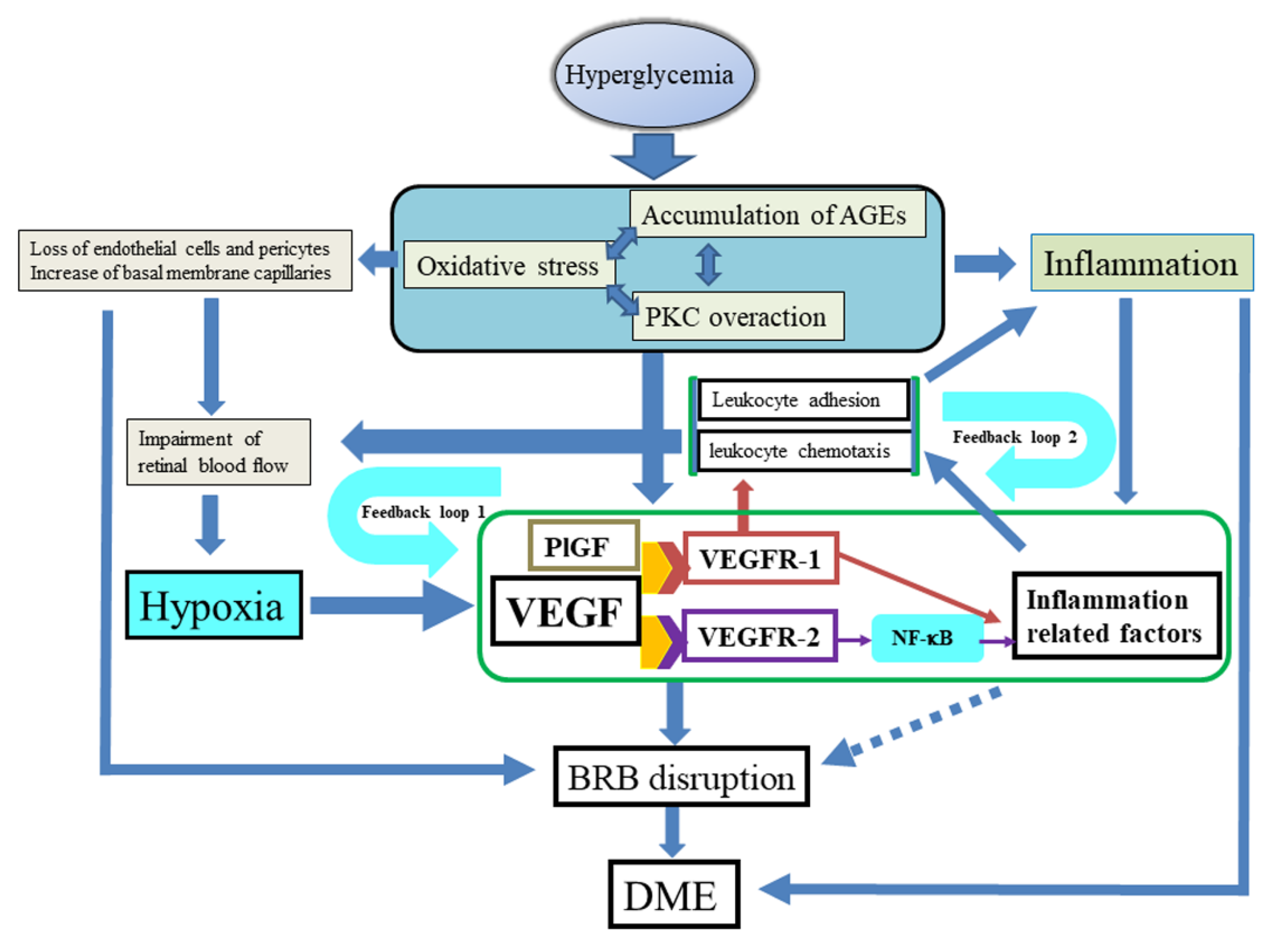
4. Hypothesis of the Mechanism of DME Proposed by the Authors
5. Therapy
6. Potential Novel Drug Targets Associated with Mediators
6.1. VEGF Designed Ankyrin Repeat Protein
6.2. Interleukin Inhibitors
6.3. Inhibitors of Adhesion Molecules
6.4. Inhibitors of Multiple Growth Factors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antcliff, R.J.; Marshall, J. The pathogenesis of edema in diabetic maculopathy. Semin. Ophthalmol. 1999, 14, 223–232. [Google Scholar] [CrossRef]
- Coscas, G.; Cunha-Vaz, J.; Soubrane, G. Macular edema: Definition and basic concepts. Dev. Ophthalmol. 2017, 58, 1–10. [Google Scholar]
- Early Treatment Diabetic Retinopathy Study Research Group. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Arch. Ophthalmol. 1985, 103, 1796–1806. [Google Scholar] [CrossRef]
- Lewis, H.; Abrams, G.W.; Blumenkranz, M.S.; Campo, R.V. Vitrectomy for diabetic macular traction and edema associated with posterior hyaloidal traction. Ophthalmology 1992, 99, 753–759. [Google Scholar] [CrossRef]
- Tachi, N.; Ogino, N. Vitrectomy for diffuse macular edema in cases of diabetic retinopathy. Am. J. Ophthalmol 1996, 122, 258–260. [Google Scholar] [CrossRef]
- Jonas, J.B.; Söfker, A. Intraocular injection of crystalline cortisone as adjunctive treatment of diabetic macular edema. Am. J. Ophthalmol 2001, 132, 425–427. [Google Scholar] [CrossRef]
- Funatsu, H.; Yamashita, H.; Noma, H.; Mimura, T.; Yamashita, T.; Hori, S. Increased levels of vascular endothelial growth factor and interleukin-6 in the aqueous humor of diabetics with macular edema. Am. J. Ophthalmol 2002, 133, 70–77. [Google Scholar] [CrossRef]
- Chun, D.W.; Heier, J.S.; Topping, T.M.; Duker, J.S.; Bankert, J.M. A pilot study of multiple intravitreal injections of ranibizumab in patients with center-involving clinically significant diabetic macular edema. Ophthalmology 2006, 113, 1706–1712. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Tatlipinar, S.; Shah, S.M.; Haller, J.A.; Quinlan, E.; Sung, J.; Zimmer-Galler, I.; Do, D.V.; Campochiaro, P.A. Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am. J. Ophthalmol. 2006, 142, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; Glassman, A.R.; Ayala, A.R.; Jampol, L.M.; Aiello, L.P.; Antoszyk, A.N.; Arnold-Bush, B.; Baker, C.W.; Bressler, N.M.; Browning, D.J.; et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N. Engl. J. Med. 2015, 372, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Aoki, S.; Ohnishi, K.; Yasuda, T.; Kawano, K.; Tsukada, Y. Increased serum levels of advanced glycation end-products and diabetic complications. Diabetes Res. Clin. Pract. 1998, 41, 131–137. [Google Scholar] [CrossRef]
- Warboys, C.M.; Fraser, P.A. Hyperglycemia attenuates acute permeability response to advanced glycation end products in retinal microvasculature. Microvasc. Res. 2010, 80, 174–176. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Cukiernik, M.; Hileeto, D.; Evans, T.; Chen, S. Role of vasoactive factors in the pathogenesis of early changes in diabetic retinopathy. Diabetes Metab. Res. Rev. 2000, 16, 393–407. [Google Scholar] [CrossRef]
- Cheung, C.M.; Vania, M.; Ang, M.; Chee, S.P.; Li, J. Comparison of aqueous humor cytokine and chemokine levels in diabetic patients with and without retinopathy. Mol. Vis. 2012, 18, 830–837. [Google Scholar]
- Cunha-Vaz, J. Diabetic macular edema. Eur. J. Ophthalmol. 1998, 8, 127–130. [Google Scholar] [CrossRef]
- Staurenghi, G.; Sadda, S.; Chakravarthy, U.; Spaide, R.F. Proposed lexicon for anatomic landmarks in normal posterior segment spectral-domain optical coherence tomography: The IN•OCT consensus. Ophthalmology 2014, 121, 1572–1578. [Google Scholar] [CrossRef]
- Guney, S.; Schuler, A.; Ott, A.; Hoschele, S.; Zugel, S.; Baloglu, E.; Bartsch, P.; Mairbaurl, H. Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+-K+-ATPase and epithelial Na+ channels. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1332–L1338. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stitt, A.W.; Lois, N.; Medina, R.J.; Adamson, P.; Curtis, T.M. Advances in our understanding of diabetic retinopathy. Clin. Sci. 2013, 125, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Li, Y.; Zhang, D.; Gee, S.; Crosson, C.; Ma, J. Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett. 2001, 489, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Arjamaa, O.; Nikinmaa, M. Oxygen-dependent diseases in the retina: Role of hypoxia-inducible factors. Exp. Eye Res. 2006, 83, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Wang, Y. Intravitreous vascular endothelial growth factor and hypoxia-inducible factor 1a in patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 2009, 148, 883–889. [Google Scholar] [CrossRef]
- Aiello, L.P.; Northrup, J.M.; Keyt, B.A.; Takagi, H.; Iwamoto, M.A. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch. Ophthalmol. 1995, 113, 1538–1544. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Hollinger, L.A.; Wolpert, E.B.; Gardner, T.W. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem. 1999, 274, 23463–23467. [Google Scholar]
- Murata, T.; Ishibashi, T.; Khalil, A.; Hata, Y.; Yoshikawa, H.; Inomata, H. Vascular endothelial growth factor plays a role in hyperpermeability of diabetic retinal vessels. Ophthalmic Res. 1995, 27, 48–52. [Google Scholar] [CrossRef]
- Murata, T.; Nakagawa, K.; Khalil, A.; Ishibashi, T.; Inomata, H.; Sueishi, K. The relation between expression of vascular endothelial growth factor and breakdown of the blood-retinal barrier in diabetic rat retinas. Lab. Investig. 1996, 74, 819–825. [Google Scholar]
- Lang, G.E. Diabetic macular edema. Ophthalmologica 2012, 1, 21–29. [Google Scholar] [CrossRef]
- Tolentino, M.J.; McLeod, D.S.; Taomoto, M.; Otsuji, T.; Adamis, A.P.; Lutty, G.A. Pathologic features of vascular endothelial growth factor-induced retinopathy in the nonhuman primate. Am. J. Ophthalmol. 2002, 133, 373–385. [Google Scholar] [CrossRef]
- Funatsu, H.; Yamashita, H.; Ikeda, T.; Mimura, T.; Eguchi, S.; Hori, S. Vitreous levels of interleukin-6 and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2003, 110, 1690–1696. [Google Scholar] [CrossRef]
- Shimura, M.; Yasuda, K.; Motohashi, R.; Kotake, O.; Noma, H. Aqueous cytokine and growth factor levels indicate response to ranibizumab for diabetic macular oedema. Br. J. Ophthalmol. 2017, 101, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Noma, H.; Mimura, T.; Eguchi, S.; Hori, S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 2009, 116, 73–79. [Google Scholar] [CrossRef]
- Xia, P.; Aiello, L.P.; Ishii, H.; Jiang, Z.Y.; Park, D.J.; Robinson, G.S.; Takagi, H.; Newsome, W.P.; Jirousek, M.R.; King, G.L. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J. Clin. Investig. 1996, 98, 2018–2026. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Venema, V.J.; Gu, X.; Venema, R.C.; Marrero, M.B.; Caldwell, R.B. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J. Biol. Chem. 1999, 274, 25130–25135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.W.; Mayo, L.D.; Dunbar, J.D.; Kessler, K.M.; Baerwald, M.R.; Jaffe, E.A.; Wang, D.; Warren, R.S.; Donner, D.B. Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J. Biol. Chem. 2000, 275, 5096–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noma, H.; Mimura, T.; Yasuda, K.; Motohashi, R.; Kotake, O.; Shimura, M. Aqueous humor levels of soluble vascular endothelial growth factor receptor and inflammatory factors in diabetic macular edema. Ophthalmologica 2017, 238, 81–88. [Google Scholar] [CrossRef]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [Green Version]
- Hornig, C.; Barleon, B.; Ahmad, S.; Vuorela, P.; Ahmed, A.; Weich, H.A. Release and complex formation of soluble VEGFR-1 from endothelial cells and biological fluids. Lab. Investig. 2000, 80, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J. Biochem. Mol. Biol. 2006, 39, 469–478. [Google Scholar] [CrossRef]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Falco, S.; Gigante, B.; Persico, M.G. Structure and function of placental growth factor. Trends Cardiovasc. Med. 2002, 12, 241–246. [Google Scholar] [CrossRef]
- Clauss, M.; Weich, H.; Breier, G.; Knies, U.; Rockl, W.; Waltenberger, J.; Risau, W. The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J. Biol. Chem. 1996, 271, 17629–17634. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Kiba, A.; Sagara, H.; Hara, T.; Shibuya, M. VEGFR-2-specific ligand VEGF-E induces non-edematous hyper-vascularization in mice. Biochem. Biophys. Res. Commun. 2003, 301, 371–377. [Google Scholar] [CrossRef]
- Murakami, M.; Iwai, S.; Hiratsuka, S.; Yamauchi, M.; Nakamura, K.; Iwakura, Y.; Shibuya, M. Signaling of vascular endothelial growth factor receptor-1 tyrosine kinase promotes rheumatoid arthritis through activation of monocytes/macrophages. Blood 2006, 108, 1849–1856. [Google Scholar] [CrossRef]
- Ledebur, H.C.; Parks, T.P. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J. Biol. Chem. 1995, 270, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [Green Version]
- Marumo, T.; Schini-Kerth, V.B.; Fisslthaler, B.; Busse, R. Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-kappaB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation 1997, 96, 2361–2367. [Google Scholar] [CrossRef]
- Ishida, S.; Usui, T.; Yamashiro, K.; Kaji, Y.; Ahmed, E.; Carrasquillo, K.G.; Amano, S.; Hida, T.; Oguchi, Y.; Adamis, A.P. VEGF164 is proinflammatory in the diabetic retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2155–2162. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; McGuire, P.G.; Rangasamy, S. Diabetic macular edema: Pathophysiology and novel therapeutic targets. Ophthalmology 2015, 122, 1375–1394. [Google Scholar] [CrossRef] [PubMed]
- Maturi, R.K.; Glassman, A.R.; Liu, D.; Beck, R.W.; Bhavsar, A.R.; Bressler, N.M.; Jampol, L.M.; Melia, M.; Punjabi, O.S.; Salehi-Had, H.; et al. Effect of adding dexamethasone to continued ranibizumab treatment in patients with persistent diabetic macular edema: A DRCR network phase 2 randomized clinical trial. JAMA Ophthalmol. 2018, 136, 29–38. [Google Scholar] [CrossRef]
- Bressler, S.B.; Ayala, A.R.; Bressler, N.M.; Melia, M.; Qin, H.; Ferris, F.L., 3rd; Flaxel, C.J.; Friedman, S.M.; Glassman, A.R.; Jampol, L.M.; et al. Persistent macular thickening after ranibizumab treatment for diabetic macular edema with vision impairment. JAMA Ophthalmol. 2016, 134, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef]
- Hernández, C.; Fonollosa, A.; García-Ramírez, M.; Higuera, M.; Catalán, R.; Miralles, A.; García-Arumí, J.; Simó, R. Erythropoietin is expressed in the human retina and it is highly elevated in the vitreous fluid of patients with diabetic macular edema. Diabetes Care 2006, 29, 2028–2033. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, T.; Sonoda, K.H.; Sugahara, M.; Mochizuki, Y.; Enaida, H.; Oshima, Y.; Ueno, A.; Hata, Y.; Yoshida, H.; Ishibashi, T. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS ONE 2009, 4, e8158. [Google Scholar] [CrossRef]
- Ando, R.; Noda, K.; Namba, S.; Saito, W.; Kanda, A.; Ishida, S. Aqueous humour levels of placental growth factor in diabetic retinopathy. Acta Ophthalmol. 2014, 92, e245–e246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ramírez, M.; Hernández, C.; Ruiz-Meana, M.; Villarroel, M.; Corraliza, L.; García-Dorado, D.; Simó, R. Erythropoietin protects retinal pigment epithelial cells against the increase of permeability induced by diabetic conditions: Essential role of JAK2/ PI3K signaling. Cell Signal. 2011, 23, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xie, H.; Yang, Q.; Yang, Y.; Li, W.; Tian, H.; Lu, L.; Wang, F.; Xu, J.Y.; Gao, F.; et al. Erythropoietin protects outer blood-retinal barrier in experimental diabetic retinopathy by up-regulating ZO-1 and occludin. Clin. Exp. Ophthalmol. 2019, 47, 1182–1197. [Google Scholar] [CrossRef]
- Grunwald, J.E.; Brucker, A.J.; Grunwald, S.E.; Riva, C.E. Retinal hemodynamics in proliferative diabetic retinopathy. A laser Doppler velocimetry study. Invest. Ophthalmol Vis. Sci. 1993, 34, 66–71. [Google Scholar]
- Arend, O.; Remky, A.; Harris, A.; Bertram, B.; Reim, M.; Wolf, S. Macular microcirculation in cystoid maculopathy of diabetic patients. Br. J. Ophthalmol. 1995, 79, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadonosono, K.; Itoh, N.; Ohno, S. Perifoveal microcirculation before and after vitrectomy for diabetic cystoid macular edema. Am. J. Ophthalmol. 2000, 130, 740–744. [Google Scholar] [CrossRef]
- Funatsu, H.; Sakata, K.; Harino, S.; Okuzawa, Y.; Noma, H.; Hori, S. Tracing method in the assessment of retinal capillary blood flow velocity by fluorescein angiography with scanning laser ophthalmoscope. Jpn. J. Ophthalmol. 2006, 50, 25–32. [Google Scholar] [CrossRef]
- Tanaka, T.; Muraoka, K.; Shimizu, K. Fluorescein fundus angiography with scanning laser ophthalmoscope. Visibility of leukocytes and platelets in perifoveal capillaries. Ophthalmology 1991, 98, 1824–1829. [Google Scholar] [CrossRef]
- Sakata, K.; Funatsu, H.; Harino, S.; Noma, H.; Hori, S. Relationship between macular microcirculation and progression of diabetic macular edema. Ophthalmology 2006, 113, 1385–1391. [Google Scholar] [CrossRef]
- Mizui, T.; Noma, H.; Yasuda, K.; Kanemaki, T.; Goto, H.; Shimura, M. Intravitreal ranibizumab reduced ocular blood flow and aqueous cytokine levels and improved retinal morphology in patients with diabetic macular edema. Sci. Rep. 2020, 10, 21713. [Google Scholar] [CrossRef]
- DiSalvo, J.; Bayne, M.L.; Conn, G.; Kwok, P.W.; Trivedi, P.G.; Soderman, D.D.; Palisi, T.M.; Sullivan, K.A.; Thomas, K.A. Purification and characterization of a naturally occurring vascular endothelial growth factor.placenta growth factor heterodimer. J. Biol. Chem. 1995, 270, 7717–7723. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar] [CrossRef]
- Olofsson, B.; Korpelainen, E.; Pepper, M.S.; Mandriota, S.J.; Aase, K.; Kumar, V.; Gunji, Y.; Jeltsch, M.M.; Shibuya, M.; Alitalo, K.; et al. Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11709–11714. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, N.; de Kozak, Y.; Normand, N.; Courtois, Y.; Jeanny, J.C.; Benezra, D.; Behar-Cohen, F. PlGF-1 and VEGFR-1 pathway regulation of the external epithelial hemato-ocular barrier. A model for retinal edema. Ophthalmic Res. 2008, 40, 203–207. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Ao, P.; Boynton, A.L. Rapid disruption of gap junctional communication and phosphorylation of connexin43 by platelet-derived growth factor in T51B rat liver epithelial cells expressing platelet-derived growth factor receptor. J. Cell Physiol. 1998, 174, 66–77. [Google Scholar] [CrossRef]
- Mamer, S.B.; Chen, S.; Weddell, J.C.; Palasz, A.; Wittenkeller, A.; Kumar, M.; Imoukhuede, P.I. Discovery of high-affinity PDGF-VEGFR Interactions: Redefining RTK dynamics. Sci. Rep. 2017, 7, 16439. [Google Scholar] [CrossRef] [Green Version]
- Dawson, D.W.; Volpert, O.V.; Gillis, P.; Crawford, S.E.; Xu, H.; Benedict, W.; Bouck, N.P. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science 1999, 285, 245–248. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Ocular neovascularization. J. Mol. Med. 2013, 91, 311–321. [Google Scholar] [CrossRef]
- Praidou, A.; Papakonstantinou, E.; Androudi, S.; Georgiadis, N.; Karakiulakis, G.; Dimitrakos, S. Vitreous and serum levels of vascular endothelial growth factor and platelet-derived growth factor and their correlation in patients with non-proliferative diabetic retinopathy and clinically significant macula oedema. Acta. Ophthalmol. 2011, 89, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Maruo, N.; Morita, I.; Shirao, M.; Murota, S. IL-6 increases endothelial permeability in vitro. Endocrinology 1992, 131, 710–714. [Google Scholar]
- Yan, S.F.; Tritto, I.; Pinsky, D.; Liao, H.; Huang, J.; Fuller, G.; Brett, J.; May, L.; Stern, D. Induction of interleukin 6 (IL-6) by hypoxia in vascular cells. Central role of the binding site for nuclear factor-IL-6. J. Biol. Chem. 1995, 270, 11463–11471. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.H.; Schlidt, S.A.; Chandel, N.S.; Hynes, K.L.; Schumacker, P.T.; Gewertz, B.L. Endothelial permeability and IL-6 production during hypoxia: Role of ROS in signal transduction. Am. J. Physiol. 1999, 277, L1057–L1065. [Google Scholar] [CrossRef]
- Pearlstein, D.P.; Ali, M.H.; Mungai, P.T.; Hynes, K.L.; Gewertz, B.L.; Schumacker, P.T. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arter. Thromb. Vasc. Biol. 2002, 22, 566–573. [Google Scholar] [CrossRef]
- Wu, J.; Zhong, Y.; Yue, S.; Yang, K.; Zhang, G.; Chen, L.; Liu, L. Aqueous humor mediator and cytokine aberrations in diabetic retinopathy and diabetic macular edema: A systematic review and meta-analysis. Dis. Markers 2019, 23, 6928524. [Google Scholar] [CrossRef] [Green Version]
- Karakurum, M.; Shreeniwas, R.; Chen, J.; Pinsky, D.; Yan, S.D.; Anderson, M.; Sunouchi, K.; Major, J.; Hamilton, T.; Kuwabara, K.; et al. Hypoxic induction of interleukin-8 gene expression in human endothelial cells. J. Clin. Investig. 1994, 93, 1564–1570. [Google Scholar] [CrossRef] [Green Version]
- Shono, T.; Ono, M.; Izumi, H.; Jimi, S.I.; Matsushima, K.; Okamoto, T.; Kohno, K.; Kuwano, M. Involvement of the transcription factor NF-kappaB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol. Cell Biol. 1996, 16, 4231–4239. [Google Scholar] [CrossRef] [Green Version]
- Moyer, K.E.; Saggers, G.C.; Allison, G.M.; Mackay, D.R.; Ehrlich, H.P. Effects of interleukin-8 on granulation tissue maturation. J. Cell Physiol. 2002, 193, 173–179. [Google Scholar] [CrossRef]
- Yu, H.; Huang, X.; Ma, Y.; Gao, M.; Wang, O.; Gao, T.; Shen, Y.; Liu, X. Interleukin-8 regulates endothelial permeability by down-regulation of tight junction but not dependent on integrins induced focal adhesions. Int. J. Biol. Sci. 2013, 9, 966–979. [Google Scholar] [CrossRef]
- Kwon, J.W.; Jee, D. Aqueous humor cytokine levels in patients with diabetic macular edema refractory to anti-VEGF treatment. PLoS ONE 2018, 13, e0203408. [Google Scholar]
- Chen, Y.L.; Chang, Y.J.; Jiang, M.J. Monocyte chemotactic protein-1 gene and protein expression in atherogenesis of hypercholesterolemic rabbits. Atherosclerosis 1999, 143, 115–123. [Google Scholar] [CrossRef]
- Chen, P.; Shibata, M.; Zidovetzki, R.; Fisher, M.; Zlokovic, B.V.; Hofman, F.M. Endothelin-1 and monocyte chemoattractant protein-1 modulation in ischemia and human brain-derived endothelial cell cultures. J. Neuroimmunol. 2001, 116, 62–73. [Google Scholar] [CrossRef]
- Lee, P.C.; Ho, I.C.; Lee, T.C. Oxidative stress mediates sodium arsenite-induced expression of heme oxygenase-1, monocyte chemoattractant protein-1, and interleukin-6 in vascular smooth muscle cells. Toxicol Sci. 2005, 85, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Keep, R.F.; Kunkel, S.L.; Andjelkovic, A.V. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: Signaling via Rho and Rho kinase. J. Cell Sci. 2003, 116, 4615–4628. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Liu, M.T.; Lei, H.Y.; Liu, C.C.; Wu, J.M.; Tung, Y.C.; Lin, Y.S.; Yeh, T.M.; Chen, S.H.; Liu, H.S. MCP-1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J. Gen. Virol. 2006, 87, 3623–3630. [Google Scholar] [CrossRef]
- Abraham, J.R.; Wykoff, C.C.; Arepalli, S.; Lunasco, L.; Yu, H.J.; Hu, M.; Reese, J.; Srivastava, S.K.; Brown, D.M.; Ehlers, J.P. Aqueous Cytokine Expression and Higher Order OCT Biomarkers: Assessment of the Anatomic-Biologic Bridge in the IMAGINE DME Study. Am. J. Ophthalmol. 2021, 222, 328–339. [Google Scholar] [CrossRef]
- Feldman, E.D.; Weinreich, D.M.; Carroll, N.M.; Burness, M.L.; Feldman, A.L.; Turner, E.; Xu, H.; Alexander, H.R., Jr. Interferon gamma-inducible protein 10 selectively inhibits proliferation and induces apoptosis in endothelial cells. Ann. Surg. Oncol. 2006, 13, 125–133. [Google Scholar] [CrossRef]
- Bodnar, R.J.; Yates, C.C.; Wells, A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ. Res. 2006, 98, 617–625. [Google Scholar] [CrossRef]
- Elner, S.G.; Elner, V.M.; Pavilack, M.A.; Todd, R.F., 3rd; Mayo-Bond, L.; Franklin, W.A.; Strieter, R.M.; Kunkel, S.L.; Huber, A.R. Modulation and function of intercellular adhesion molecule-1 (CD54) on human retinal pigment epithelial cells. Lab. Investig. 1992, 66, 200–211. [Google Scholar] [PubMed]
- Nishiwaki, A.; Ueda, T.; Ugawa, S.; Shimada, S.; Ogura, Y. Upregulation of P-selectin and intercellular adhesion molecule-1 after retinal ischemia-reperfusion injury. Invest. Ophthalmol Vis. Sci. 2003, 44, 4931–4935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, F.; Kiryu, J.; Miyamoto, K.; Nishijima, K.; Miyahara, S.; Katsuta, H.; Tamura, H.; Honda, Y. In vivo evaluation of retinal injury after transient ischemia in hypertensive rats. Hypertension 2004, 43, 1098–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, D.S.; Lefer, D.J.; Merges, C.; Lutty, G.A. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am. J. Pathol. 1995, 147, 642–653. [Google Scholar]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Moromizato, Y.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am. J. Pathol. 2000, 156, 1733–1739. [Google Scholar] [CrossRef] [Green Version]
- Joussen, A.M.; Poulaki, V.; Qin, W.; Kirchhof, B.; Mitsiades, N.; Wiegand, S.J.; Rudge, J.; Yancopoulos, G.D.; Adamis, A.P. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am. J. Pathol. 2002, 160, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.R.; Prescott, E.; Shelke, N.B.; Trivedi, R.; Thomas, P.; Struble, C.; Gadek, T.; O’Neill, C.A.; Kompella, U.B. Delivery of SAR 1118 to the retina via ophthalmic drops and its effectiveness in a rat streptozotocin (STZ) model of diabetic retinopathy (DR). Invest. Ophthalmol Vis. Sci. 2010, 51, 5198–5204. [Google Scholar] [CrossRef] [Green Version]
- Hillier, R.J.; Ojaimi, E.; Wong, D.T.; Mak, M.Y.K.; Berger, A.R.; Kohly, R.P.; Kertes, P.J.; Forooghian, F.; Boyd, S.R.; Eng, K.; et al. Aqueous humor cytokine levels and anatomic response to intravitreal ranibizumab in diabetic macular edema. JAMA Ophthalmol. 2018, 136, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Noma, H.; Mimura, T.; Yasuda, K.; Shimura, M. Role of inflammation in diabetic macular edema. Ophthalmologica 2014, 232, 127–135. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Wykoff, C.C.; Shapiro, H.; Rubio, R.G.; Ehrlich, J.S. Neutralization of vascular endothelial growth factor slows progression of retinal nonperfusion in patients with diabetic macular edema. Ophthalmology 2014, 121, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Elman, M.J.; Aiello, L.P.; Beck, R.W.; Bressler, N.M.; Bressler, S.B.; Edwards, A.R.; Ferris, F.L., 3rd; Friedman, S.M.; Glassman, A.R.; Miller, K.M.; et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 2010, 117, 1064–1077.e1035. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.; Bandello, F.; Schmidt-Erfurth, U.; Lang, G.E.; Massin, P.; Schlingemann, R.O.; Sutter, F.; Simader, C.; Burian, G.; Gerstner, O.; et al. The RESTORE study: Ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology 2011, 118, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.D.; Brown, D.M.; Marcus, D.M.; Boyer, D.S.; Patel, S.; Feiner, L.; Gibson, A.; Sy, J.; Rundle, A.C.; Hopkins, J.J.; et al. Ranibizumab for diabetic macular edema: Results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology 2012, 119, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Korobelnik, J.F.; Do, D.V.; Schmidt-Erfurth, U.; Boyer, D.S.; Holz, F.G.; Heier, J.S.; Midena, E.; Kaiser, P.K.; Terasaki, H.; Marcus, D.M.; et al. Intravitreal aflibercept for diabetic macular edema. Ophthalmology 2014, 121, 2247–2254. [Google Scholar] [CrossRef]
- Ishibashi, T.; Li, X.; Koh, A.; Lai, T.Y.; Lee, F.L.; Lee, W.K.; Ma, Z.; Ohji, M.; Tan, N.; Cha, S.B.; et al. The REVEAL study: Ranibizumab monotherapy or combined with laser versus laser monotherapy in Asian patients with diabetic macular edema. Ophthalmology 2015, 122, 1402–1415. [Google Scholar] [CrossRef]
- Boyer, D.S.; Nguyen, Q.D.; Brown, D.M.; Basu, K.; Ehrlich, J.S. Outcomes with As-needed ranibizumab after initial monthly therapy: Long-term outcomes of the phase III RIDE and RISE trials. Ophthalmology 2015, 122, 2504–2513. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Erfurth, U.; Lang, G.E.; Holz, F.G.; Schlingemann, R.O.; Lanzetta, P.; Massin, P.; Gerstner, O.; Bouazza, A.S.; Shen, H.; Osborne, A.; et al. Three-year outcomes of individualized ranibizumab treatment in patients with diabetic macular edema: The RESTORE extension study. Ophthalmology 2014, 121, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Elman, M.J.; Ayala, A.; Bressler, N.M.; Browning, D.; Flaxel, C.J.; Glassman, A.R.; Jampol, L.M.; Stone, T.W. Intravitreal Ranibizumab for diabetic macular edema with prompt versus deferred laser treatment: 5-year randomized trial results. Ophthalmology 2015, 122, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Imazeki, M.; Noma, H.; Yasuda, K.; Motohashi, R.; Goto, H.; Shimura, M. Anti-VEGF therapy reduces inflammation in diabetic macular edema. Ophthalmic Res. 2021, 64, 43–49. [Google Scholar]
- Arimura, N.; Otsuka, H.; Yamakiri, K.; Sonoda, Y.; Nakao, S.; Noda, Y.; Hashiguchi, T.; Maruyama, I.; Sakamoto, T. Vitreous mediators after intravitreal bevacizumab or triamcinolone acetonide in eyes with proliferative diabetic retinopathy. Ophthalmology 2009, 116, 921–926. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Hafiz, G.; Mir, T.A.; Scott, A.W.; Zimmer-Galler, I.; Shah, S.M.; Wenick, A.S.; Brady, C.J.; Han, I.; He, L.; et al. Pro-permeability factors in diabetic macular edema. The Diabetic macular edema treated with ozurdex trial. Am. J. Ophthalmol. 2016, 168, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elman, M.J.; Bressler, N.M.; Qin, H.; Beck, R.W.; Ferris, F.L., 3rd; Friedman, S.M.; Glassman, A.R.; Scott, I.U.; Stockdale, C.R.; Sun, J.K. Expanded 2-year follow-up of ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 2011, 118, 609–614. [Google Scholar] [CrossRef] [Green Version]
- Shimura, M.; Yasuda, K.; Minezaki, T.; Noma, H. Reduction in the frequency of intravitreal bevacizumab administrations achieved by posterior subtenon injection of triamcinolone acetonide in patients with diffuse diabetic macular edema. Jpn. J. Ophthalmol. 2016, 60, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Bressler, S.B.; Qin, H.; Melia, M.; Bressler, N.M.; Beck, R.W.; Chan, C.K.; Grover, S.; Miller, D.G. Exploratory analysis of the effect of intravitreal ranibizumab or triamcinolone on worsening of diabetic retinopathy in a randomized clinical trial. JAMA Ophthalmol. 2013, 131, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ip, M.S.; Domalpally, A.; Sun, J.K.; Ehrlich, J.S. Long-term effects of therapy with ranibizumab on diabetic retinopathy severity and baseline risk factors for worsening retinopathy. Ophthalmology 2015, 122, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.M.; Schmidt-Erfurth, U.; Do, D.V.; Holz, F.G.; Boyer, D.S.; Midena, E.; Heier, J.S.; Terasaki, H.; Kaiser, P.K.; Marcus, D.M.; et al. Intravitreal aflibercept for diabetic macular edema: 100-week results from the VISTA and VIVID studies. Ophthalmology 2015, 122, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.K.; Wang, P.W.; Taylor, S.; Haskova, Z. Durability of diabetic retinopathy improvement with as-needed ranibizumab: Open-label extension of RIDE and RISE studies. Ophthalmology 2019, 126, 712–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, A.; Rey, P.A.; Erginay, A.; Lavia, C.; Bonnin, S.; Dupas, B.; Gaudric, A.; Tadayoni, R. Widefield OCT-angiography and fluorescein angiography assessments of nonperfusion in diabetic retinopathy and edema treated with anti-vascular endothelial growth factor. Ophthalmology 2019, 126, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Takamura, Y.; Tomomatsu, T.; Matsumura, T.; Arimura, S.; Gozawa, M.; Takihara, Y.; Inatani, M. The effect of photocoagulation in ischemic areas to prevent recurrence of diabetic macular edema after intravitreal bevacizumab injection. Investig. Ophthalmol. Vis. Sci 2014, 55, 4741–4746. [Google Scholar] [CrossRef] [Green Version]
- Campochiaro, P.A.; Channa, R.; Berger, B.B.; Heier, J.S.; Brown, D.M.; Fiedler, U.; Hepp, J.; Stumpp, M.T. Treatment of diabetic macular edema with a designed ankyrin repeat protein that binds vascular endothelial growth factor: A phase I/II study. Am. J. Ophthalmol. 2013, 155, 697–704. [Google Scholar] [CrossRef]
- Smithwick, E.; Stewart, M.W. Designed ankyrin repeat proteins: A look at their evolving use in medicine with a focus on the treatment of chorioretinal vascular disorders. Antiinflamm. Antiallergy Agents Med. Chem. 2017, 16, 33–45. [Google Scholar] [CrossRef]
- Karkhur, S.; Hasanreisoglu, M.; Vigil, E.; Halim, M.S.; Hassan, M.; Plaza, C.; Nguyen, N.V.; Afridi, R.; Tran, A.T.; Do, D.V.; et al. Interleukin-6 inhibition in the management of non-infectious uveitis and beyond. J. Ophthalmic Inflamm. Infect. 2019, 9, 19–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Morita, M.; Tojo, T.; Nagashima, A.; Moritomo, A.; Miyake, H. Novel 1H-imidazol-2-amine derivatives as potent and orally active vascular adhesion protein-1 (VAP-1) inhibitors for diabetic macular edema treatment. Bioorg. Med. Chem. 2013, 21, 3873–3881. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.D.; Sepah, Y.J.; Berger, B.; Brown, D.; Do, D.V.; Garcia-Hernandez, A.; Patel, S.; Rahhal, F.M.; Shildkrot, Y.; Renfurm, R.W. Primary outcomes of the VIDI study: Phase 2, double-masked, randomized, active-controlled study of ASP8232 for diabetic macular edema. Int. J. Retin. Vitr. 2019, 5, 19–178. [Google Scholar] [CrossRef] [Green Version]
- Sills, A.K., Jr.; Williams, J.I.; Tyler, B.M.; Epstein, D.S.; Sipos, E.P.; Davis, J.D.; McLane, M.P.; Pitchford, S.; Cheshire, K.; Gannon, F.H.; et al. Squalamine inhibits angiogenesis and solid tumor growth in vivo and perturbs embryonic vasculature. Cancer Res. 1998, 58, 2784–2792. [Google Scholar]
- Wroblewski, J.J.; Hu, A.Y. Topical squalamine 0.2% and intravitreal ranibizumab 0.5 mg as combination therapy for macular edema due to branch and central retinal vein occlusion: An open-label, randomized study. Ophthalmic. Surg. Lasers Imaging Retin. 2016, 47, 914–923. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noma, H.; Yasuda, K.; Shimura, M. Involvement of Cytokines in the Pathogenesis of Diabetic Macular Edema. Int. J. Mol. Sci. 2021, 22, 3427. https://doi.org/10.3390/ijms22073427
Noma H, Yasuda K, Shimura M. Involvement of Cytokines in the Pathogenesis of Diabetic Macular Edema. International Journal of Molecular Sciences. 2021; 22(7):3427. https://doi.org/10.3390/ijms22073427
Chicago/Turabian StyleNoma, Hidetaka, Kanako Yasuda, and Masahiko Shimura. 2021. "Involvement of Cytokines in the Pathogenesis of Diabetic Macular Edema" International Journal of Molecular Sciences 22, no. 7: 3427. https://doi.org/10.3390/ijms22073427
APA StyleNoma, H., Yasuda, K., & Shimura, M. (2021). Involvement of Cytokines in the Pathogenesis of Diabetic Macular Edema. International Journal of Molecular Sciences, 22(7), 3427. https://doi.org/10.3390/ijms22073427