
Gene Transactivation and Transrepression in MYC-Driven Cancers




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
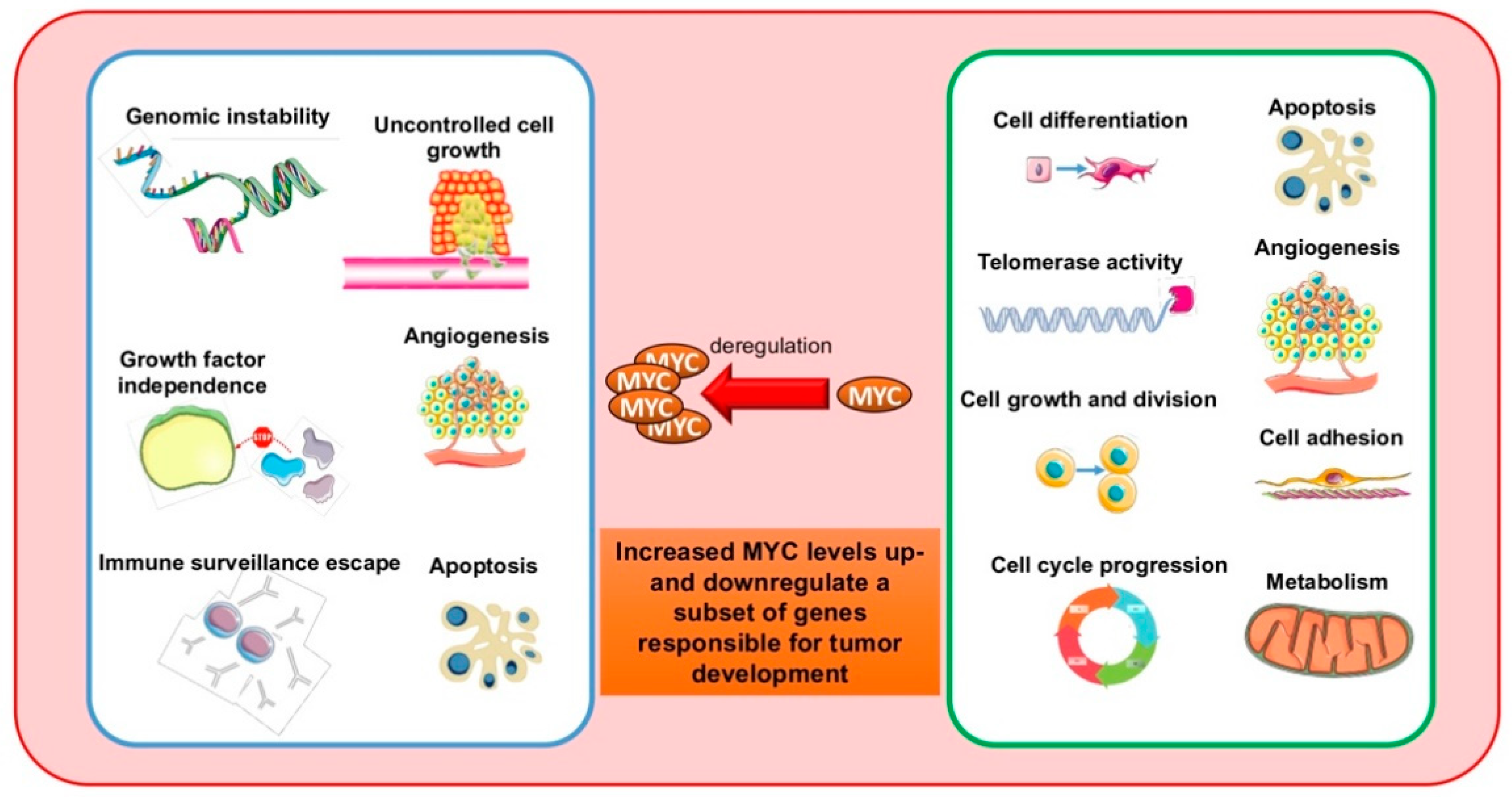
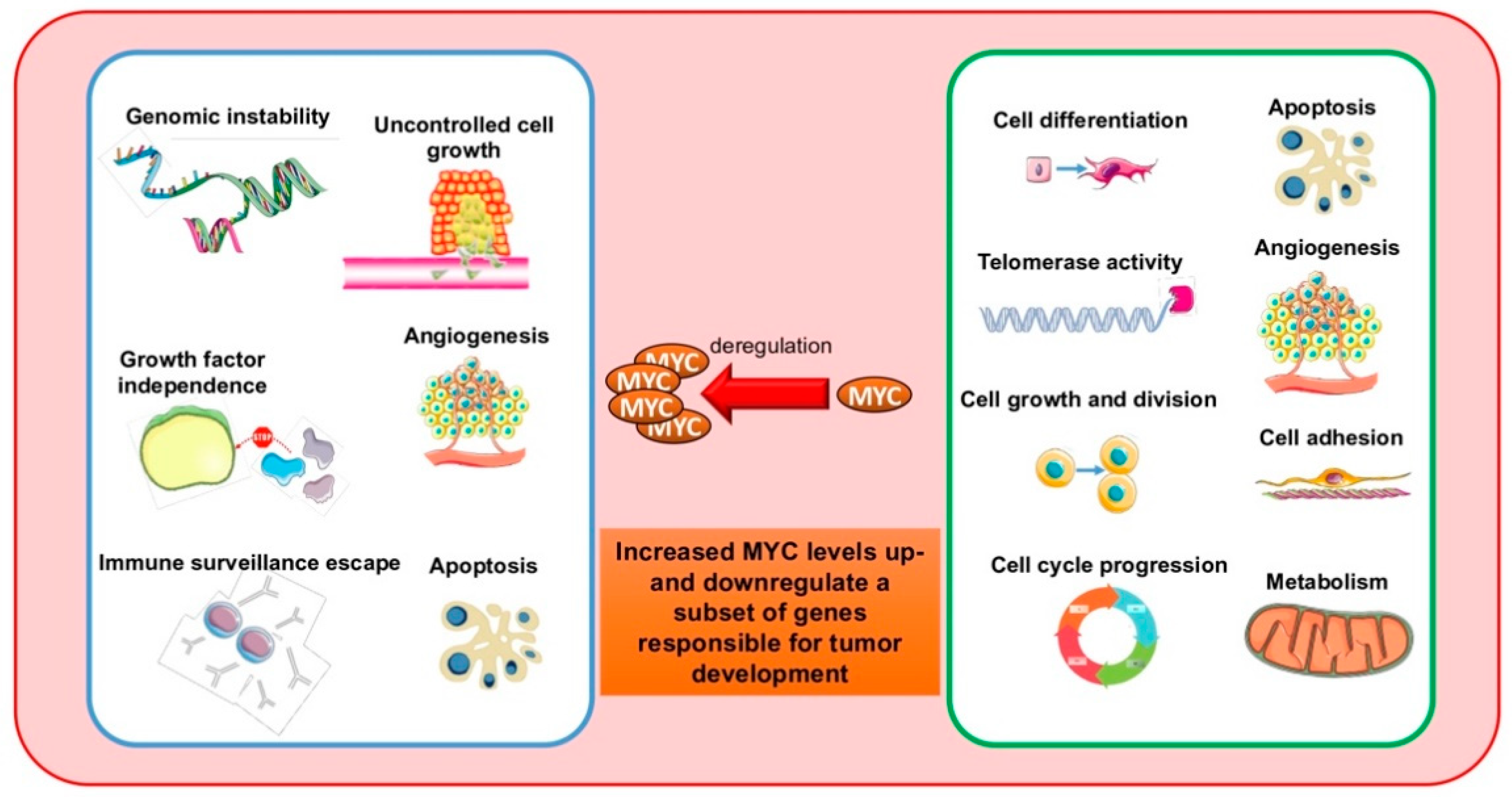
:1. Introduction
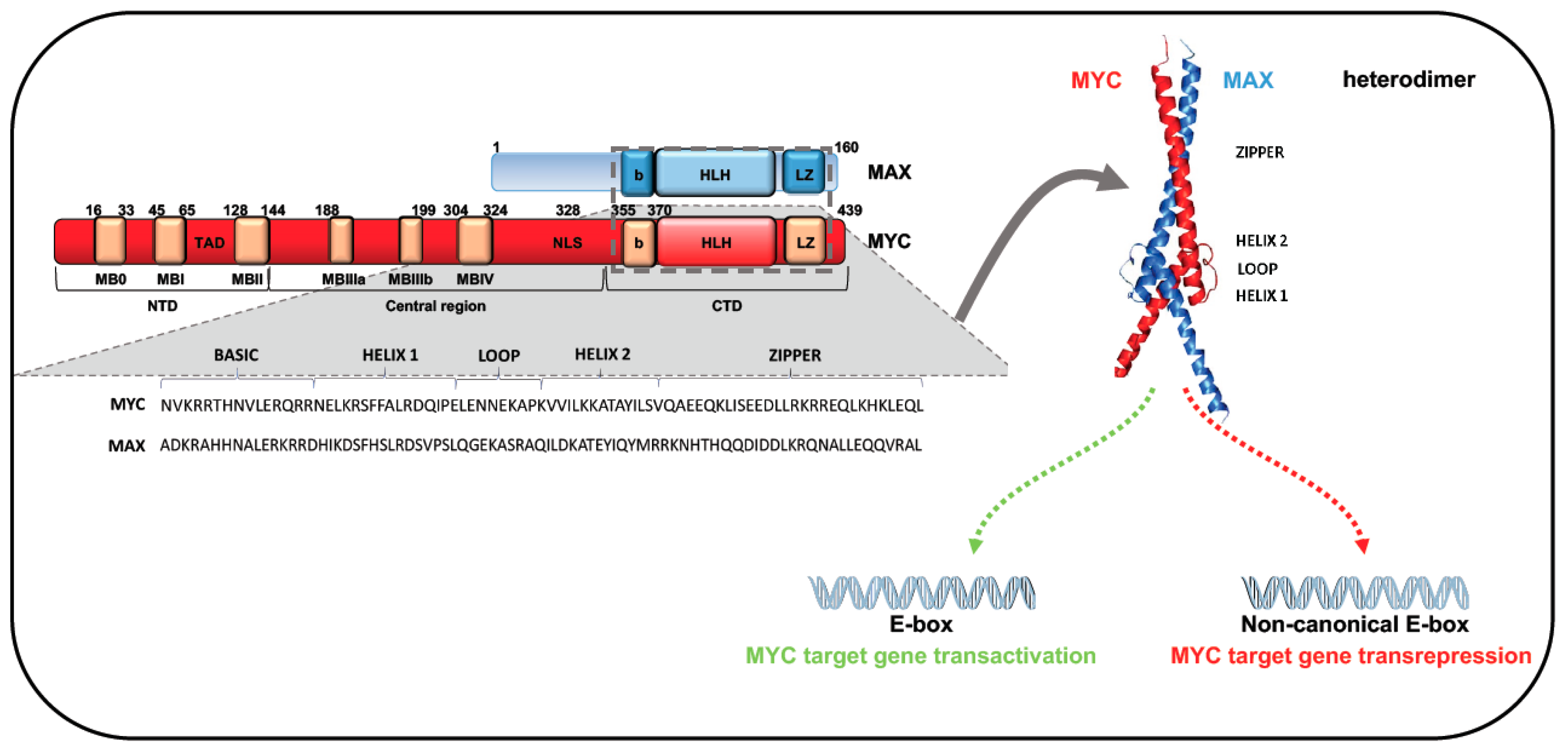
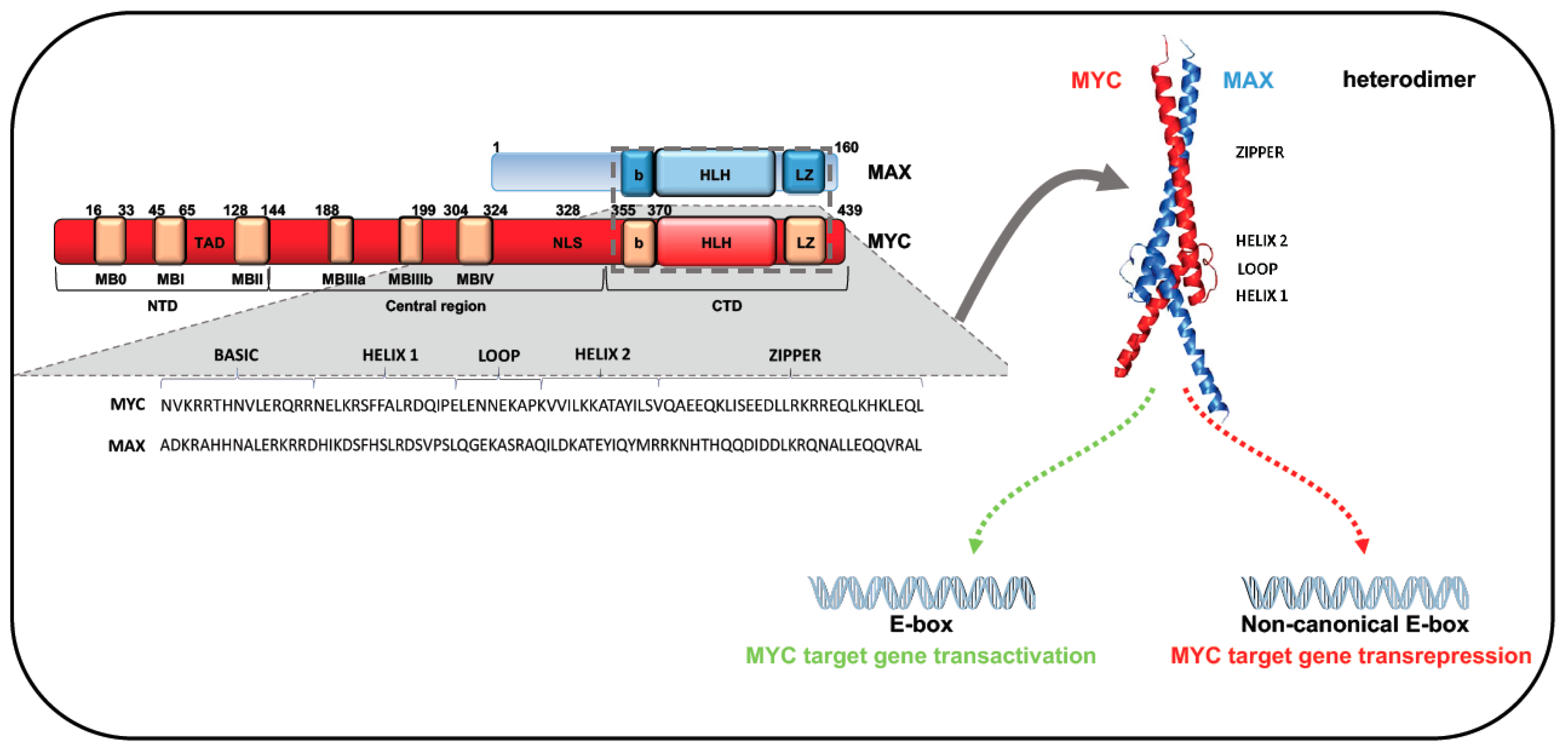
2. MYC Gene Family: Structure and Function
3. MYC in Cell Cycle
3.1. MYC-P27KIP1 Antagonism
3.2. MYC and MIZ1 Action
3.3. MYC-p53 Negative Correlation
3.4. MYC-p53 Crosstalk in Tumorigenesis
3.5. MYC/BIN1 Interaction: Cell Death Program Regulation
4. Role of MYC in the Homeostasis of Hematopoietic Stem Cells
4.1. Alterations of MYC Pathways in Lymphoma and Leukemia
4.1.1. MYC in Acute Myeloid Leukemia
4.1.2. MYC in Double-Hit Lymphoma
4.1.3. MYC in Chronic Myeloid Leukemia
4.1.4. MYC in Burkitt Lymphoma
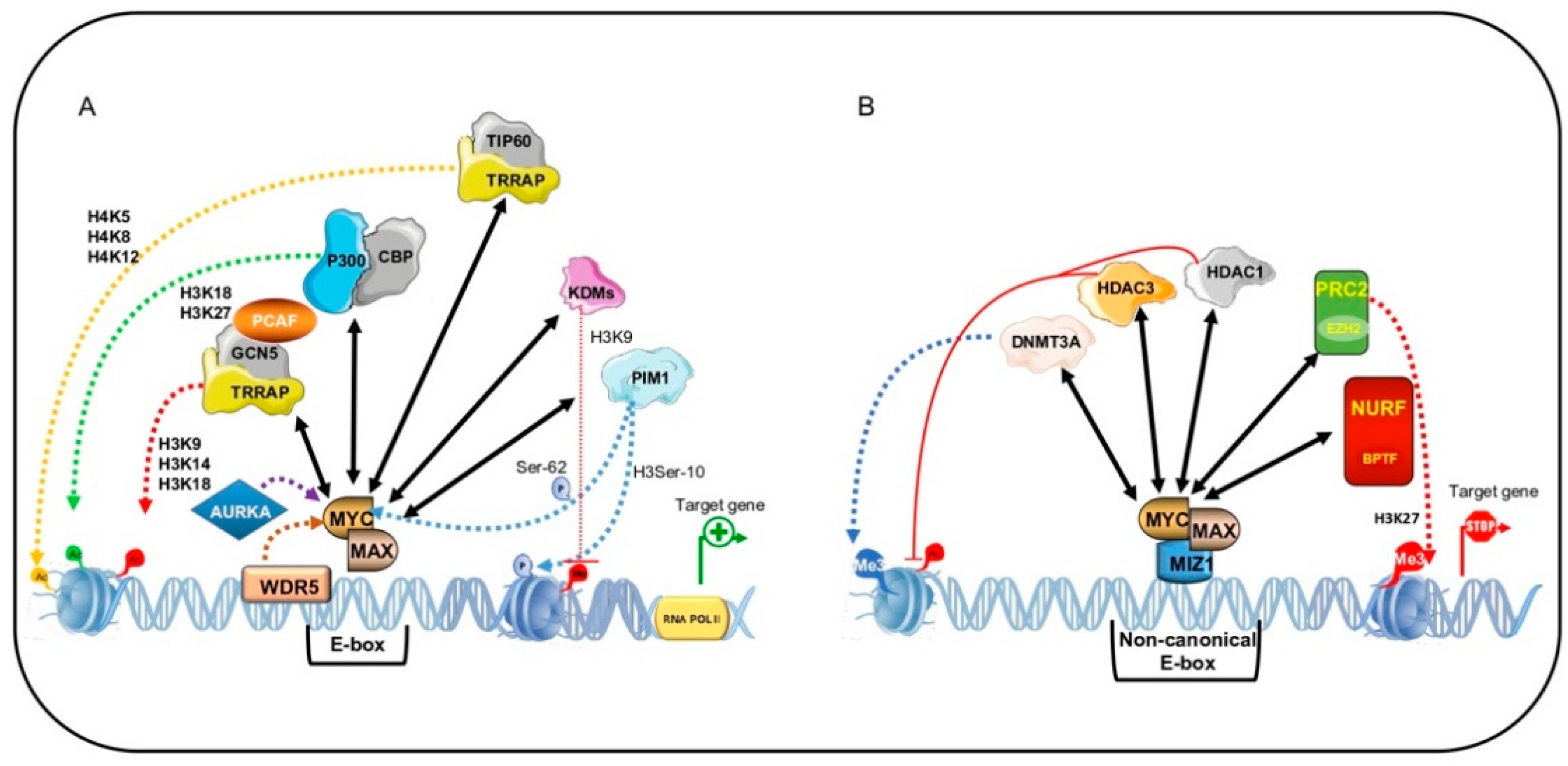
5. MYC-Mediated Transcriptional Output Regulation
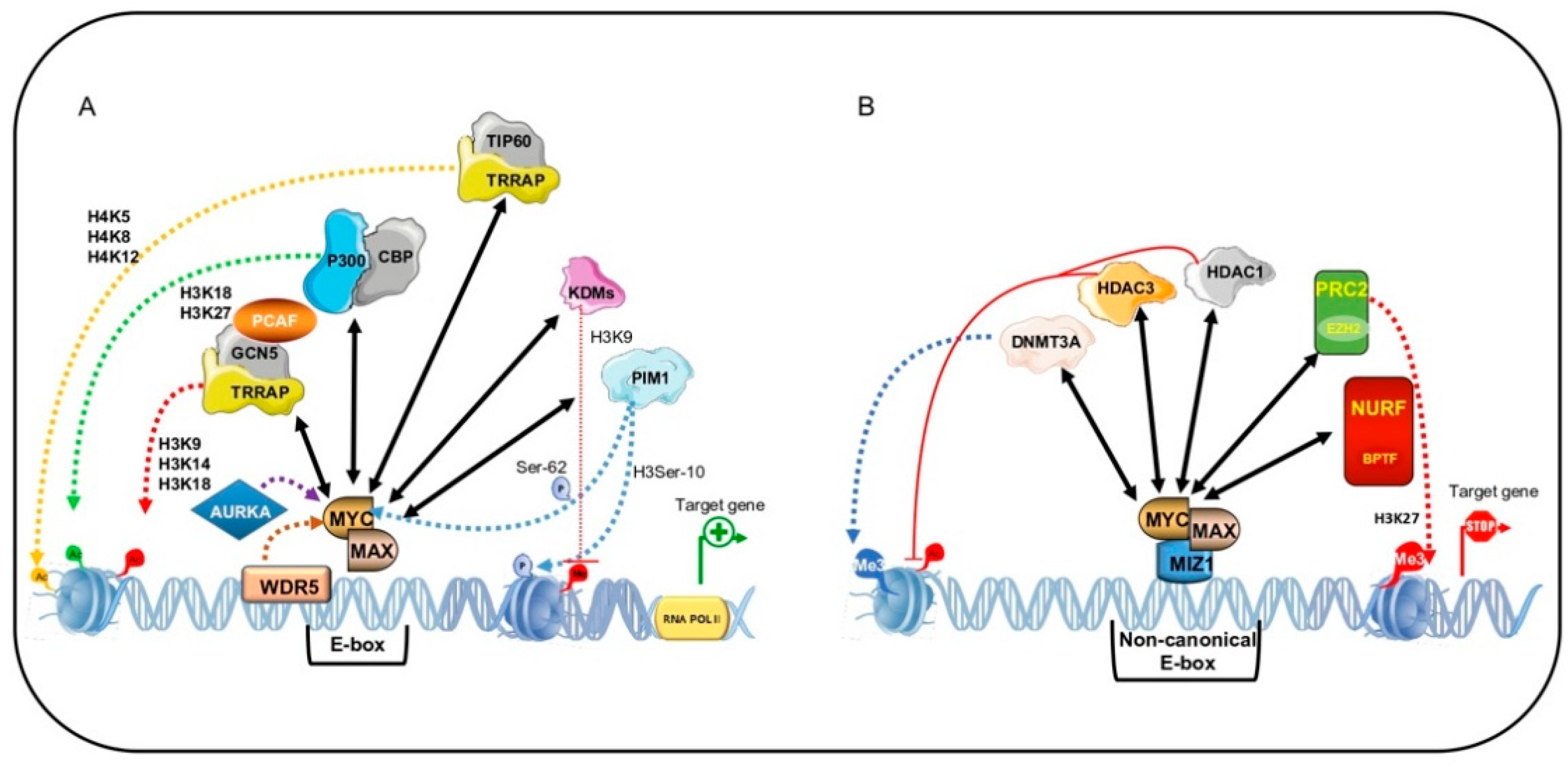
5.1. MYC-Dependent Transactivation
5.2. MYC-Dependent Transrepression
5.3. BPTF: MYC Co-Factor for Chromatin Remodeling in Human Cancer
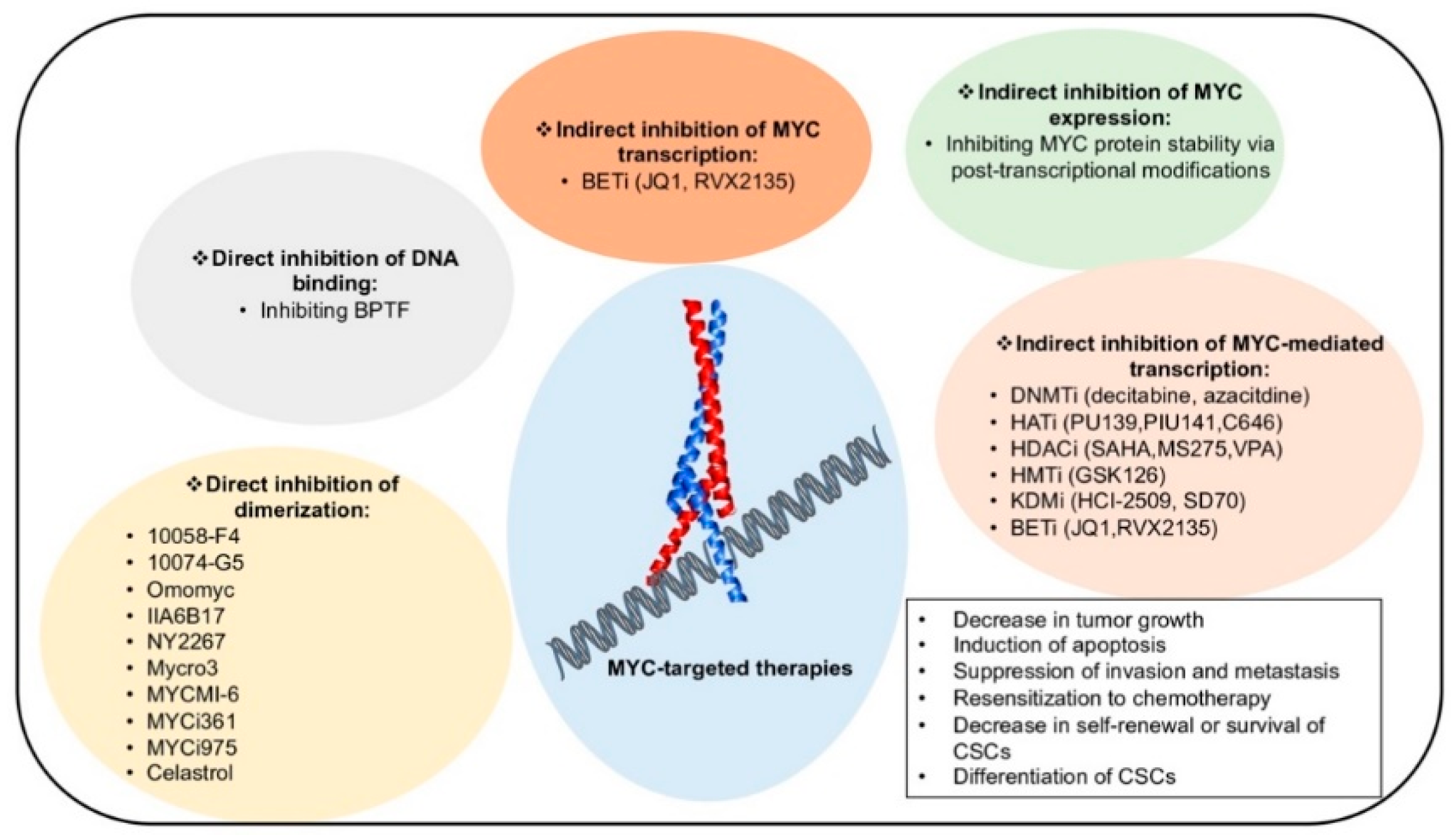
6. Therapeutic Strategies to Target MYC
6.1. Targeting Epigenetic Mechanisms Controlled by MYC
6.2. Inhibitors of MYC:MAX Heterodimerization
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duesberg, P.H.; Vogt, P.K. Avian acute leukemia viruses MC29 and MH2 share specific RNA sequences: Evidence for a second class of transforming genes. Proc. Natl. Acad. Sci. USA 1979, 76, 1633–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellon, P.; Pawson, A.; Bister, K.; Martin, G.S.; Duesberg, P.H. Specific RNA sequences and gene products of MC29 avian acute leukemia virus. Proc. Natl. Acad. Sci. USA 1978, 75, 5874–5878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finver, S.N.; Nishikura, K.; Finger, L.R.; Haluska, F.G.; Finan, J.; Nowell, P.C.; Croce, C.M. Sequence analysis of the MYC oncogene involved in the t(8;14)(q24;q11) chromosome translocation in a human leukemia T-cell line indicates that putative regulatory regions are not altered. Proc. Natl. Acad. Sci. USA 1988, 85, 3052–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef]
- McKeown, M.R.; Bradner, J.E. Therapeutic Strategies to Inhibit MYC. Cold Spring Harb. Perspect. Med. 2014, 4, a014266. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Allevato, M.; Bolotin, E.; Grossman, M.; Mane-Padros, D.; Sladek, F.M.; Martinez, E. Sequence-specific DNA binding by MYC/MAX to low-affinity non-E-box motifs. PLoS ONE 2017, 12, e0180147. [Google Scholar] [CrossRef] [Green Version]
- Muhle-Goll, C.; Gibson, T.; Schuck, P.; Schubert, D.; Nalis, D.; Nilges, M.; Pastore, A. The dimerization stability of the HLH-LZ transcription protein family is modulated by the leucine zippers: A CD and NMR study of TFEB and c-Myc. Biochemisty 1994, 33, 11296–11306. [Google Scholar] [CrossRef]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc–Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Flinn, E.M.; Busch, C.M.C.; Wright, A.P.H. myc Boxes, Which Are Conserved in myc Family Proteins, Are Signals for Protein Degradation via the Proteasome. Mol. Cell. Biol. 1998, 18, 5961–5969. [Google Scholar] [CrossRef] [Green Version]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.-K.; Wei, Y.; Shiah, Y.-J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahram, F.; Von Der Lehr, N.; Cetinkaya, C.; Larsson, L.-G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110. [Google Scholar] [CrossRef] [PubMed]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.-C.; Kang, T.-W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC–aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- McMahon, S.B.; Van Buskirk, H.A.; Dugan, K.A.; Copeland, T.D.; Cole, M.D. The Novel ATM-Related Protein TRRAP Is an Essential Cofactor for the c-Myc and E2F Oncoproteins. Cell 1998, 94, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Brough, D.E.; Hofmann, T.J.; Ellwood, K.B.; Townley, R.A.; Cole, M.D. An essential domain of the c-myc protein interacts with a nuclear factor that is also required for E1A-mediated transformation. Mol. Cell. Biol. 1995, 15, 1536–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Skp2 Regulates Myc Protein Stability and Activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar] [CrossRef]
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol. Cell Biol. 2006, 26, 4226–4239. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.R.; Adams, C.M.; Wang, J.; Weissmiller, A.M.; Creighton, J.; Lorey, S.L.; Liu, Q.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. USA 2019, 116, 25260–25268. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Guo, C.; Das, S.K.; Chow, C.C.; Batchelor, E.; Simons, S.S.; Levens, D. Dissecting transcriptional amplification by MYC. eLife 2020, 9, e52483. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A Large Scale Genetic Analysis of c-Myc-regulated Gene Expression Patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.-S.; Peterson, A.L.; Castellani, G.; Sedivy, J.M.; Neretti, N. Kinetic profiling of the c-Myc transcriptome and bioinformatic analysis of repressed gene promoters. Cell Cycle 2011, 10, 2184–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.R.; Sears, R.; Nuckolls, F.; Leone, G.; Nevins, J.R. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol. Cell Biol. 2000, 20, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Sears, R.; Huang, E.; Rempel, R.; Nuckolls, F.; Park, C.-H.; Giangrande, P.; Wu, L.; Saavedra, H.I.; Field, S.J.; et al. Myc Requires Distinct E2F Activities to Induce S Phase and Apoptosis. Mol. Cell 2001, 8, 105–113. [Google Scholar] [CrossRef]
- Sears, R.; Ohtani, K.; Nevins, J.R. Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol. 1997, 17, 5227–5235. [Google Scholar] [CrossRef] [Green Version]
- García-Gutiérrez, L.; Bretones, G.; Molina, E.; Arechaga, I.; Symonds, C.; Acosta, J.C.; Blanco, R.; Fernández, A.; Alonso, L.; Sicinski, P.; et al. Myc stimulates cell cycle progression through the activation of Cdk1 and phosphorylation of p27. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Lee, M.-H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massagué, J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef]
- Coats, S.; Flanagan, W.M.; Nourse, J.; Roberts, J.M. Requirement of p27Kip1 for Restriction Point Control of the Fibroblast Cell Cycle. Science 1996, 272, 877–880. [Google Scholar] [CrossRef]
- Perez-Roger, I.; Kim, S.; Griffiths, B.; Sewing, A.; Land, H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J. 1999, 18, 5310–5320. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, C.; Thieke, K.; Maier, A.; Saffrich, R.; Hanley-Hyde, J.; Ansorge, W.; Reed, S.; Sicinski, P.; Bartek, J.; Eilers, M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999, 18, 5321–5333. [Google Scholar] [CrossRef]
- Bretones, G.; Acosta, J.C.; Caraballo, J.M.; Ferrándiz, N.; Gómez-Casares, M.T.; Albajar, M.; Blanco, R.; Ruiz, P.; Hung, W.-C.; Albero, M.P.; et al. SKP2 Oncogene Is a Direct MYC Target Gene and MYC Down-regulates p27KIP1 through SKP2 in Human Leukemia Cells*. J. Biol. Chem. 2011, 286, 9815–9825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y. [A report of investigations on an outbreak of type B paretyphoid fever]. Zhonghua Liu Xing Bing Xue Za Zhi 1990, 11, 288–290. [Google Scholar] [PubMed]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massagué, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.B.; Eilers, M.; Massagué, J. TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.-V.; Massagué, J. Myc suppression of the p21Cip1 Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nat. Cell Biol. 2002, 419, 729–734. [Google Scholar] [CrossRef]
- Wu, S.; Çetinkaya, C.; Muñoz-Alonso, M.J.; Von Der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; León, J.; Larsson, L.-G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.; Mo, Y.Y. p53 and c-myc: How does the cell balance "yin" and "yang"? Cell Cycle 2009, 8, 1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.-S.; Jin, Y.; Gallegos, J.R.; Lu, H. Balance of Yin and Yang: Ubiquitylation-Mediated Regulation of p53 and c-Myc. Neoplasia 2006, 8, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. p53-Dependent Transcriptional Repression of c-myc Is Required for G1 Cell Cycle Arrest. Mol. Cell. Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef]
- Levy, N.; Yonish-Rouach, E.; Oren, M.; Kimchi, A. Complementation by wild-type p53 of interleukin-6 effects on M1 cells: Induction of cell cycle exit and cooperativity with c-myc suppression. Mol. Cell. Biol. 1993, 13, 7942–7952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragimov, N.; Krauskopf, A.; Navot, N.; Rotter, V.; Oren, M.; Aloni, Y. Wild-type but not mutant p53 can repress transcription initiation in vitro by interfering with the binding of basal transcription factors to the TATA motif. Oncogene 1993, 8, 1183–1193. [Google Scholar]
- Schultz, M.; Kühne, W. The problems of early diagnosis of diffuse malignant mesothelioma from the pathologo-anatomic view. Zentralblatt Pathol. 1992, 138, 85–90. [Google Scholar]
- Yu, L.; Yu, T.-T.; Young, K.H. Cross-talk between Myc and p53 in B-cell lymphomas. Chronic Dis. Transl. Med. 2019, 5, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hussain, S.P.; De Fromentel, C.C.; Hainaut, P.; Harris, C.C. TP53 mutation spectra and load: A tool for generating hypotheses on the etiology of cancer. IARC Sci. Public 2004, 157, 247–270. [Google Scholar]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenz, T.; Kreuz, M.; Fuge, M.; Klapper, W.; Horn, H.; Staiger, A.M.; Winter, D.; Helfrich, H.; Huellein, J.; Hansmann, M.; et al. TP53 mutation and survival in aggressive B cell lymphoma. Int. J. Cancer 2017, 141, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulz, P.; Heitzer, E.; Speicher, M.R. Co-occurrence of MYC amplification and TP53 mutations in human cancer. Nat. Genet. 2016, 48, 104–106. [Google Scholar] [CrossRef]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Huang, X.; Zhang, Y.; Tang, Y.; Butler, N.; Kim, J.; Guessous, F.; Schiff, D.; Mandell, J.; Abounader, R. A novel PTEN/mutant p53/c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia 2013, 15, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The Tumor Suppressor p53 Regulates Polarity of Self-Renewing Divisions in Mammary Stem Cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624–638. [Google Scholar] [CrossRef] [Green Version]
- Korac, P.; Dotlic, S.; Matulic, M.; Zajc Petranovic, M.; Dominis, M. Role of MYC in B Cell Lymphomagenesis. Genes 2017, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- Askew, D.S.; Ashmun, R.A.; Simmons, B.C.; Cleveland, J.L. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991, 6, 1915–1922. [Google Scholar]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Shi, Y.; Glynn, J.; Guilbert, L.; Cotter, T.; Bissonnette, R.; Green, D. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 1992, 257, 212–214. [Google Scholar] [CrossRef]
- Bhatia, K.; Huppi, K.; Spangler, G.; Siwarski, D.; Iyer, R.; Magrath, I. Point mutations in the c–Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat. Genet. 1993, 5, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Sander, C.A.; Clark, H.M.; Dolezal, M.V.; Jaffe, E.S.; Raffeld, M. Clustered mutations in the second exon of the MYC gene in sporadic Burkitt’s lymphoma. Oncogene 1993, 8, 2741–2748. [Google Scholar] [PubMed]
- Sakamuro, D.; Elliott, K.J.; Wechsler-Reya, R.; Prendergast, G.C. BIN1 is a novel MYC–interacting protein with features of a tumour suppressor. Nat. Genet. 1996, 14, 69–77. [Google Scholar] [CrossRef]
- Elliott, K.; Ge, K.; Du, W.; Prendergast, G.C. The c-Myc-interacting adaptor protein Bin1 activates a caspase-independent cell death program. Oncogene 2000, 19, 4669–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda-Lucena, A.; Ho, C.S.; Mao, D.Y.; Sheng, Y.; Laister, R.C.; Muhandiram, R.; Lu, Y.; Seet, B.T.; Katz, S.; Szyperski, T.; et al. A Structure-based Model of the c-Myc/Bin1 Protein Interaction Shows Alternative Splicing of Bin1 and c-Myc Phosphorylation are Key Binding Determinants. J. Mol. Biol. 2005, 351, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. c-MYC Suppresses BIN1 to Release Poly(ADP-Ribose) Polymerase 1: A Mechanism by Which Cancer Cells Acquire Cisplatin Resistance. Sci. Signal. 2011, 4, ra19. [Google Scholar] [CrossRef]
- Ganesan, S. MYC, PARP1, and Chemoresistance: BIN There, Done That? Sci. Signal. 2011, 4, pe15. [Google Scholar] [CrossRef]
- Wang, J.; Jia, Y.; Zhao, S.; Zhang, X.; Wang, X.; Han, X.; Wang, Y.; Ma, M.; Shi, J.; Liu, L. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene 2017, 36, 6235–6243. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Tumbar, T.; Guasch, G. Socializing with the neighbors: Stem cells and their niche. Cell 2004, 116, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.-C.; Knabenhans, C.; Macdonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004, 18, 2747–2763. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Niu, C.; Breslin, P.; Tang, M.; Zhang, S.; Wei, W.; Kini, A.R.; Paner, G.P.; Alkan, S.; Morris, S.W.; et al. c-Myc–mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Blood 2009, 114, 2097–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, J.A.; Cole, M.D. Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell differentiation but not commitment. Nat. Cell Biol. 1986, 320, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W.; et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef]
- Li, L.; Osdal, T.; Ho, Y.; Chun, S.; McDonald, T.; Agarwal, P.; Lin, A.; Chu, S.; Qi, J.; Hsieh, Y.-T.; et al. SIRT1 Activation by a c-MYC Oncogenic Network Promotes the Maintenance and Drug Resistance of Human FLT3-ITD Acute Myeloid Leukemia Stem Cells. Cell Stem. Cell 2014, 15, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Basit, F.; Andersson, M.; Hultquist, A. The Myc/Max/Mxd Network Is a Target of Mutated Flt3 Signaling in Hematopoietic Stem Cells in Flt3-ITD-Induced Myeloproliferative Disease. Stem. Cells Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Tian, X.; Pelton, A.; Shahsafaei, A.; Dorfman, D.M. Differential expression of enhancer of zeste homolog 2 (EZH2) protein in small cell and aggressive B-cell non-Hodgkin lymphomas and differential regulation of EZH2 expression by p-ERK1/2 and MYC in aggressive B-cell lymphomas. Mod. Pathol. 2016, 29, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, B.; Iosue, I.; Damas, N.D.; Mangiavacchi, A.; Chiaretti, S.; Messina, M.; Padula, F.; Guarini, A.; Bozzoni, I.; Fazi, F.; et al. Critical Role of c-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes Cancer 2011, 2, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle-López, A.; León, J. MYC oncogene in myeloid neoplasias. Clin. Transl. Oncol. 2012, 15, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.; Jayne, S.; Kennedy, B.; Miall, F.; Aukema, S.M.; Siebert, R.; Wagner, S.D.; Dyer, M.J.S. A Double Hit CD10-Negative B-Cell Lymphoma with t(3;8)(q27;q24) Leading to Juxtaposition of the BCL6 and MYC Loci Associated with Good Clinical Outcome. Case Rep. Hematol. 2014, 2014, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; Isobe, Y.; Uemura, Y.; Nishio, Y.; Sakai, H.; Kato, M.; Otsubo, K.; Hoshikawa, M.; Takagi, M.; Miura, I. De novo acute lymphoblastic leukemia-like disease of high grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements: A case report and literature review. BMC Clin. Pathol. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, C.H.; Tirado, C.A. Two Double-hit Lymphomas Cases: A Molecular Cytogenetic Approach. J. Assoc. Genet. Technol. 2018, 44, 141–145. [Google Scholar]
- Li, W.; Gupta, S.K.; Han, W.; Kundson, R.A.; Nelson, S.; Knutson, D.; Greipp, P.T.; Elsawa, S.F.; Sotomayor, E.M.; Gupta, M. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, D.S.; Amer, A.M.; Mudawi, D.; Nawaz, Z.; Alkuwari, E.; Al Sabbagh, A.; Ibrahim, F.; Yassin, M.A. Chronic Myeloid Leukemia with cryptic Philadelphia translocation and extramedullary B-lymphoid blast phase as an initial presentation. Acta Biomed. 2018, 89, 38–44. [Google Scholar] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Magistroni, V.; Piazza, R.; Citterio, S.; Mezzatesta, C.; Khandelwal, P.; Pirola, A.; Gambacorti-Passerini, C. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol. Cancer 2015, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Lin, H.; Sun, T.; Arlinghaus, R.B. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 2002, 21, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic Mechanisms in Burkitt Lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282. [Google Scholar] [CrossRef] [Green Version]
- De Falco, G.; Ambrosio, M.R.; Fuligni, F.; Onnis, A.; Bellan, C.; Rocca, B.J.; Navari, M.; Etebari, M.; Mundo, L.; Gazaneo, S.; et al. Burkitt lymphoma beyond MYC translocation: N-MYC and DNA methyltransferases dysregulation. BMC Cancer 2015, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- God, J.M.; Cameron, C.; Figueroa, J.; Amria, S.; Hossain, A.; Kempkes, B.; Bornkamm, G.W.; Stuart, R.K.; Blum, J.S.; Haque, A. Elevation of c-MYC Disrupts HLA Class II–Mediated Immune Recognition of Human B Cell Tumors. J. Immunol. 2015, 194, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepel, J.B.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Huang, S.; Zhang, H.; Alikhan, M.; et al. Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Giulino-Roth, L.; Van Besien, H.J.; Dalton, T.; Totonchy, J.E.; Rodina, A.; Taldone, T.; Bolaender, A.; Erdjument-Bromage, H.; Sadek, J.; Chadburn, A.; et al. Inhibition of Hsp90 Suppresses PI3K/AKT/mTOR Signaling and Has Antitumor Activity in Burkitt Lymphoma. Mol. Cancer Ther. 2017, 16, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Poole, C.J.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; Van Riggelen, J. Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition. Cancers 2018, 10, 448. [Google Scholar] [CrossRef] [Green Version]
- Zeller, K.I.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. An integrated database of genes responsive to the Myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003, 4, R69. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; De Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nat. Cell Biol. 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Olio, V.D.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006, 8, 764–770. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad Network and the Transcriptional Control of Cell Behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Frank, S.R.; Schroeder, M.; Fernandez, P.; Taubert, S.; Amati, B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001, 15, 2069–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Kidder, B.L.; Yang, J.; Palmer, S. Stat3 and c-Myc Genome-Wide Promoter Occupancy in Embryonic Stem Cells. PLoS ONE 2008, 3, e3932. [Google Scholar] [CrossRef] [Green Version]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Zeller, K.I.; Zhao, X.; Lee, C.W.H.; Chiu, K.P.; Yao, F.; Yustein, J.T.; Ooi, H.S.; Orlov, Y.L.; Shahab, A.; Yong, H.C.; et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17834–17839. [Google Scholar] [CrossRef] [Green Version]
- Arabi, A.; Wu, S.; Ridderstråle, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlén, S.; Hydbring, P.; Söderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct activation of RNA polymerase III transcription by c-Myc. Nat. Cell Biol. 2003, 421, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; Von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. eLife 2016, 5, e15161. [Google Scholar] [CrossRef] [PubMed]
- Eberhardy, S.R.; Farnham, P.J. c-Myc Mediates Activation of the cad Promoter via a Post-RNA Polymerase II Recruitment Mechanism. J. Biol. Chem. 2001, 276, 48562–48571. [Google Scholar] [CrossRef] [Green Version]
- Eberhardy, S.R.; Farnham, P.J. Myc Recruits P-TEFb to Mediate the Final Step in the Transcriptional Activation of the cad Promoter. J. Biol. Chem. 2002, 277, 40156–40162. [Google Scholar] [CrossRef] [Green Version]
- Baluapuri, A.; Hofstetter, J.; Stankovic, N.D.; Endres, T.; Bhandare, P.; Vos, S.M.; Adhikari, B.; Schwarz, J.D.; Narain, A.; Vogt, M.; et al. MYC Recruits SPT5 to RNA Polymerase II to Promote Processive Transcription Elongation. Mol. Cell 2019, 74, 674–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhar, M.; Ebert, A.; Neumann, T.; Umkehrer, C.; Jude, J.; Wieshofer, C.; Rescheneder, P.; Lipp, J.J.; Herzog, V.A.; Reichholf, B.; et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 2018, 360, 800–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabò, A.; Amati, B. BRD4 and MYC—clarifying regulatory specificity. Science 2018, 360, 713–714. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Wood, M.A.; Cole, M.D. THE Essential Cofactor TRRAP Recruits the Histone Acetyltransferase hGCN5 to c-Myc. Mol. Cell. Biol. 2000, 20, 556–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.R.; Parisi, T.; Taubert, S.; Fernandez, P.; Fuchs, M.; Chan, H.; Livingston, D.M.; Amati, B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003, 4, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Gerber, J.; Drapkin, R.; Sif, S.; Ikura, T.; Ogryzko, V.; Lane, W.S.; Nakatani, Y.; Livingston, D.M. The p400 Complex Is an Essential E1A Transformation Target. Cell 2001, 106, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, N.S.; Ramsbottom, B.A.; Gomez-Roman, N.; Marshall, L.; Cole, P.A.; White, R.J. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 14917–14922. [Google Scholar] [CrossRef] [Green Version]
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Farina, A.; Martinez, E. Dual Regulation of c-Myc by p300 via Acetylation-Dependent Control of Myc Protein Turnover and Coactivation of Myc-Induced Transcription. Mol. Cell. Biol. 2005, 25, 10220–10234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Faiola, F.; Martinez, E. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem. Biophys. Res. Commun. 2005, 336, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. 2016, 6, 4–430. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.-J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC Oncoprotein Is a Substrate of the Acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [Green Version]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; Angeles, A.D.L.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and Combinatorial Functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in Mouse Embryonic Stem Cell Identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Altahan, A.M.; Hu, D.; Wang, Y.; Cheng, P.-H.; Morton, C.L.; Qu, C.; Nathwani, A.C.; Shohet, J.M.; Fotsis, T.; et al. The Role of Histone Demethylase KDM4B in Myc Signaling in Neuroblastoma. J. Natl. Cancer Inst. 2015, 107, djv080. [Google Scholar] [CrossRef] [Green Version]
- Oliviero, S.; Zippo, A.; De Robertis, A.; Serafini, R. PIM1-dependent phosphorylation of Histone H3 at Serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Précéd. 2007, 9, 932–944. [Google Scholar] [CrossRef]
- Ivaldi, M.S.; Karam, C.S.; Corces, V.G. Phosphorylation of histone H3 at Ser10 facilitates RNA polymerase II release from promoter-proximal pausing in Drosophila. Genes Dev. 2007, 21, 2818–2831. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, (2), 241–250. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Deplus, R.; Loriot, A.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; Pelicci, P.G.; Amati, B.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2004, 24, 336–346. [Google Scholar] [CrossRef]
- Palakurthy, R.K.; Wajapeyee, N.; Santra, M.K.; Gazin, C.; Lin, L.; Gobeil, S.; Green, M.R. Epigenetic Silencing of the RASSF1A Tumor Suppressor Gene through HOXB3-Mediated Induction of DNMT3B Expression. Mol. Cell 2009, 36, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Huerta, M.; Muñoz, R.; Tapia, R.; Soto-Reyes, E.; Ramírez, L.; Recillas-Targa, F.; González-Mariscal, L.; Lopez-Bayghen, E. Cyclin D1 Is Transcriptionally Down-Regulated by ZO-2 via an E Box and the Transcription Factor c-Myc. Mol. Biol. Cell 2007, 18, 4826–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Tee, A.E.L.; Porro, A.; Smith, S.A.; Dwarte, T.; Liu, P.Y.; Iraci, N.; Sekyere, E.; Haber, M.; Norris, M.D.; et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18682–18687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Elahi, A.; Ajidahun, A.; Clark, W.; Hernandez, J.; Achille, A.; Hao, J.-H.; Seto, E.; Shibata, D. The interplay between histone deacetylases and c-Myc in the transcriptional suppression of HPP1 in colon cancer. Cancer Biol. Ther. 2014, 15, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of MYC-Mediated miR-29 by HDAC3 and EZH2 as a Therapeutic Target of Histone Modification in Aggressive B-Cell Lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, M.A.; McMahon, S.B.; Cole, M.D. An ATPase/Helicase Complex Is an Essential Cofactor for Oncogenic Transformation by c-Myc. Mol. Cell 2000, 5, 321–330. [Google Scholar] [CrossRef]
- Mao, Y.-Q.; Houry, W.A. The Role of Pontin and Reptin in Cellular Physiology and Cancer Etiology. Front. Mol. Biosci. 2017, 4, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Sandaltzopoulos, R.; Wang, H.-M.; Hamiche, A.; Ranallo, R.; Lee, K.-M.; Fu, D.; Wu, C. Dual Functions of Largest NURF Subunit NURF301 in Nucleosome Sliding and Transcription Factor Interactions. Mol. Cell 2001, 8, 531–543. [Google Scholar] [CrossRef]
- Alkhatib, S.G.; Landry, J.W. The Nucleosome Remodeling Factor. FEBS Lett. 2011, 585, 3197–3207. [Google Scholar] [CrossRef] [Green Version]
- Richart, L.; Pau, E.C.-D.S.; Río-Machín, A.; De Andrés, M.P.; Cigudosa, J.C.; Lobo, V.J.S.-A.; Real, F.X. BPTF is required for c-MYC transcriptional activity and in vivo tumorigenesis. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Höglund, A.; Nilsson, L.M.; Forshell, L.P.; MacLean, K.H.; Nilsson, J.A. Myc sensitizes p53-deficient cancer cells to the DNA-damaging effects of the DNA methyltransferase inhibitor decitabine. Blood 2009, 113, 4281–4288. [Google Scholar] [CrossRef] [Green Version]
- Guan, H.; Xie, L.; Klapproth, K.; Weitzer, C.D.; Wirth, T.; Ushmorov, A. Decitabine represses translocated MYC oncogene in Burkitt lymphoma. J. Pathol. 2013, 229, 775–783. [Google Scholar] [CrossRef]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef]
- Gajer, J.M.; Furdas, S.D.; Grunder, A.A.; Gothwal, M.; Heinicke, U.; Keller, K.M.; Colland, F.; Fulda, S.; Pahl, H.L.; Fichtner, I.; et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis 2015, 4, e137. [Google Scholar] [CrossRef] [Green Version]
- Simpson, S.; Fiches, G.; Jean, M.J.; Dieringer, M.; McGuinness, J.; John, S.P.; Shamay, M.; Desai, P.; Zhu, J.; Santoso, N.G. Inhibition of Tip60 Reduces Lytic and Latent Gene Expression of Kaposi’s Sarcoma-Associated Herpes Virus (KSHV) and Proliferation of KSHV-Infected Tumor Cells. Front. Microbiol. 2018, 9, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Liu, F.; Asai, T.; Lai, F.; Man, N.; Xu, H.; Chen, S.; Greenblatt, S.; Hamard, P.-J.; Ando, K.; et al. Loss of p300 accelerates MDS-associated leukemogenesis. Leukemia 2016, 31, 1382–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Abbondanza, C.; Martens, J.H.A.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 2017, 23, 2542–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, L.M.; Zhou, X.; Xu, W.-S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Liu, M.; Pan, J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget 2017, 8, 3396–3411. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Weston, A.; Bearrs, J.; Thode, T.; Neiss, A.; Soldi, R.; Sharma, S. Reversible lysine-specific demethylase 1 antagonist HCI-2509 inhibits growth and decreases c-MYC in castration- and docetaxel-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2016, 19, 349–357. [Google Scholar] [CrossRef]
- Yang, J.; Milasta, S.; Ashutosh, M.; Altahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting Histone Demethylases in MYC-Driven Neuroblastomas with Ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Kim, G.W.; Yoo, J.; Lee, S.W.; Jeon, Y.H.; Kim, S.Y.; Kang, H.G.; Kim, D.-H.; Chun, K.-H.; Choi, J.; et al. Histone demethylase KDM4C controls tumorigenesis of glioblastoma by epigenetically regulating p53 and c-Myc. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Shao, Q.; Kannan, A.; Lin, Z.; Stack, B.C.; Suen, J.Y.; Gao, L. BET Protein Inhibitor JQ1 Attenuates Myc-Amplified MCC Tumor Growth In Vivo. Cancer Res. 2014, 74, 7090–7102. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Z.; Wang, Z.; Wang, S.; Chen, Z.; Li, Z.; Zhang, M.; Zou, J.; Dong, B.; Gao, J.; et al. Targeting c-Myc: JQ1 as a promising option for c-Myc-amplified esophageal squamous cell carcinoma. Cancer Lett. 2018, 419, 64–74. [Google Scholar] [CrossRef]
- Li, N.; Yang, L.; Qi, X.-K.; Lin, Y.-X.; Xie, X.; He, G.-P.; Feng, Q.-S.; Liu, L.-R.; Xie, X.; Zeng, Y.-X.; et al. BET bromodomain inhibitor JQ1 preferentially suppresses EBV-positive nasopharyngeal carcinoma cells partially through repressing c-Myc. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Roderick, J.E.; Tesell, J.; Shultz, L.D.; Brehm, M.A.; Greiner, D.L.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Gutierrez, A.; Look, A.T.; et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 2014, 123, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-S.; Chen, J.-Y.; Tsai, H.-J.; Chen, T.-Y.; Hung, W.-C. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. 2015, 5, e313. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef] [Green Version]
- Berg, T. Small-molecule modulators of c-Myc/Max and Max/Max interactions. Curr. Top. Microbiol. Immunol. 2011, 348, 139–149. [Google Scholar]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Shi, J.; Yamamoto, N.; Moss, J.A.; Vogt, P.K.; Janda, K.D. A credit-card library approach for disrupting protein–protein interactions. Bioorganic Med. Chem. 2006, 14, 2660–2673. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanner, J.; Romashko, D.; Werner, D.S.; May, E.W.; Peng, Y.; Schulz, R.; Foreman, K.W.; Russo, S.; Arnold, L.D.; Pingle, M.; et al. Reversible Linkage of Two Distinct Small Molecule Inhibitors of Myc Generates a Dimeric Inhibitor with Improved Potency That Is Active in Myc Over-Expressing Cancer Cell Lines. PLoS ONE 2015, 10, e0121793. [Google Scholar] [CrossRef] [PubMed]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In Vitro Cytotoxicity and In Vivo Efficacy, Pharmacokinetics, and Metabolism of 10074-G5, a Novel Small-Molecule Inhibitor of c-Myc/Max Dimerization. J. Pharmacol. Exp. Ther. 2010, 335, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ma, X.; Jones, H.M.; Chan, L.L.-Y.; Song, F.; Zhang, W.; Bae-Jump, V.L.; Zhou, C. Evaluation of the antitumor effects of c-Myc-Max heterodimerization inhibitor 100258-F4 in ovarian cancer cells. J. Transl. Med. 2014, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kiessling, A.; Wiesinger, R.; Sperl, B.; Berg, T. Selective Inhibition of c-Myc/Max Dimerization by a Pyrazolo[1,5-a]pyrimidine. ChemMedChem 2007, 2, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Szabolcs, M.; Koul, S.; Li, Z.; Polyzos, A.; Anagnostopoulos, C.; Cournia, Z.; Tamvakopoulos, C.; Klinakis, A.; Efstratiadis, A. Therapeutic Effects of an Anti-Myc Drug on Mouse Pancreatic Cancer. J. Natl. Cancer Inst. 2014, 106, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef]
- Wang, H.; Teriete, P.; Huabo, W.; Raveendra-Panickar, D.; Pendelton, K.; Lazo, J.S.; Eiseman, J.; Holien, T.; Misund, K.; Oliynyk, G.; et al. Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 2015, 6, 32380–32395. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tatò, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scafuro, M.; Capasso, L.; Carafa, V.; Altucci, L.; Nebbioso, A. Gene Transactivation and Transrepression in MYC-Driven Cancers. Int. J. Mol. Sci. 2021, 22, 3458. https://doi.org/10.3390/ijms22073458
Scafuro M, Capasso L, Carafa V, Altucci L, Nebbioso A. Gene Transactivation and Transrepression in MYC-Driven Cancers. International Journal of Molecular Sciences. 2021; 22(7):3458. https://doi.org/10.3390/ijms22073458
Chicago/Turabian StyleScafuro, Marika, Lucia Capasso, Vincenzo Carafa, Lucia Altucci, and Angela Nebbioso. 2021. "Gene Transactivation and Transrepression in MYC-Driven Cancers" International Journal of Molecular Sciences 22, no. 7: 3458. https://doi.org/10.3390/ijms22073458
APA StyleScafuro, M., Capasso, L., Carafa, V., Altucci, L., & Nebbioso, A. (2021). Gene Transactivation and Transrepression in MYC-Driven Cancers. International Journal of Molecular Sciences, 22(7), 3458. https://doi.org/10.3390/ijms22073458