
Yersiniabactin Siderophore of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Is Involved in Autophagy Activation in Host Cells

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
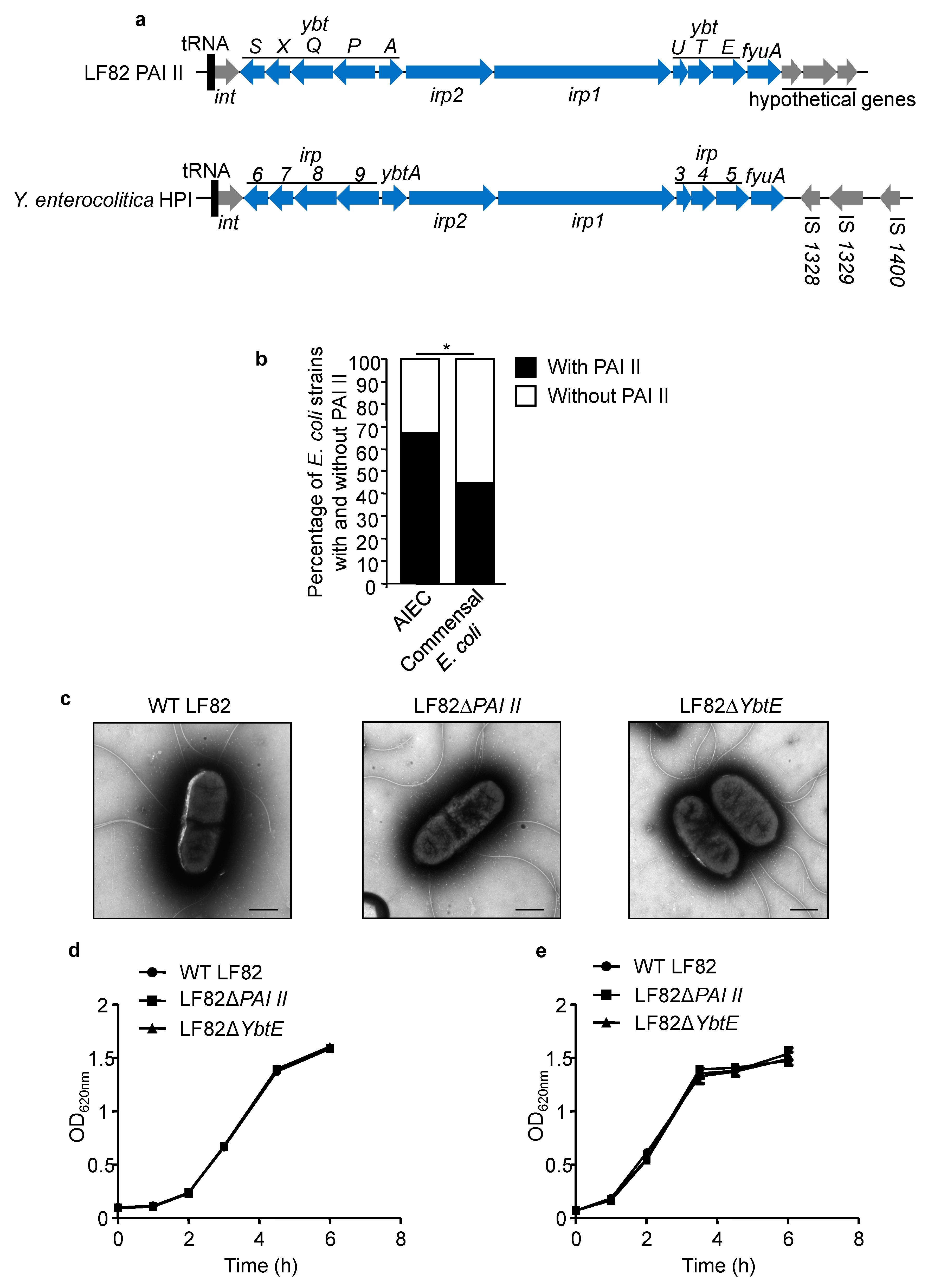
2.1. PAI II Is Over-Represented in AIEC Strains
2.2. Loss of Functional Yersiniabactin Does Not Alter AIEC Morphology and Growth
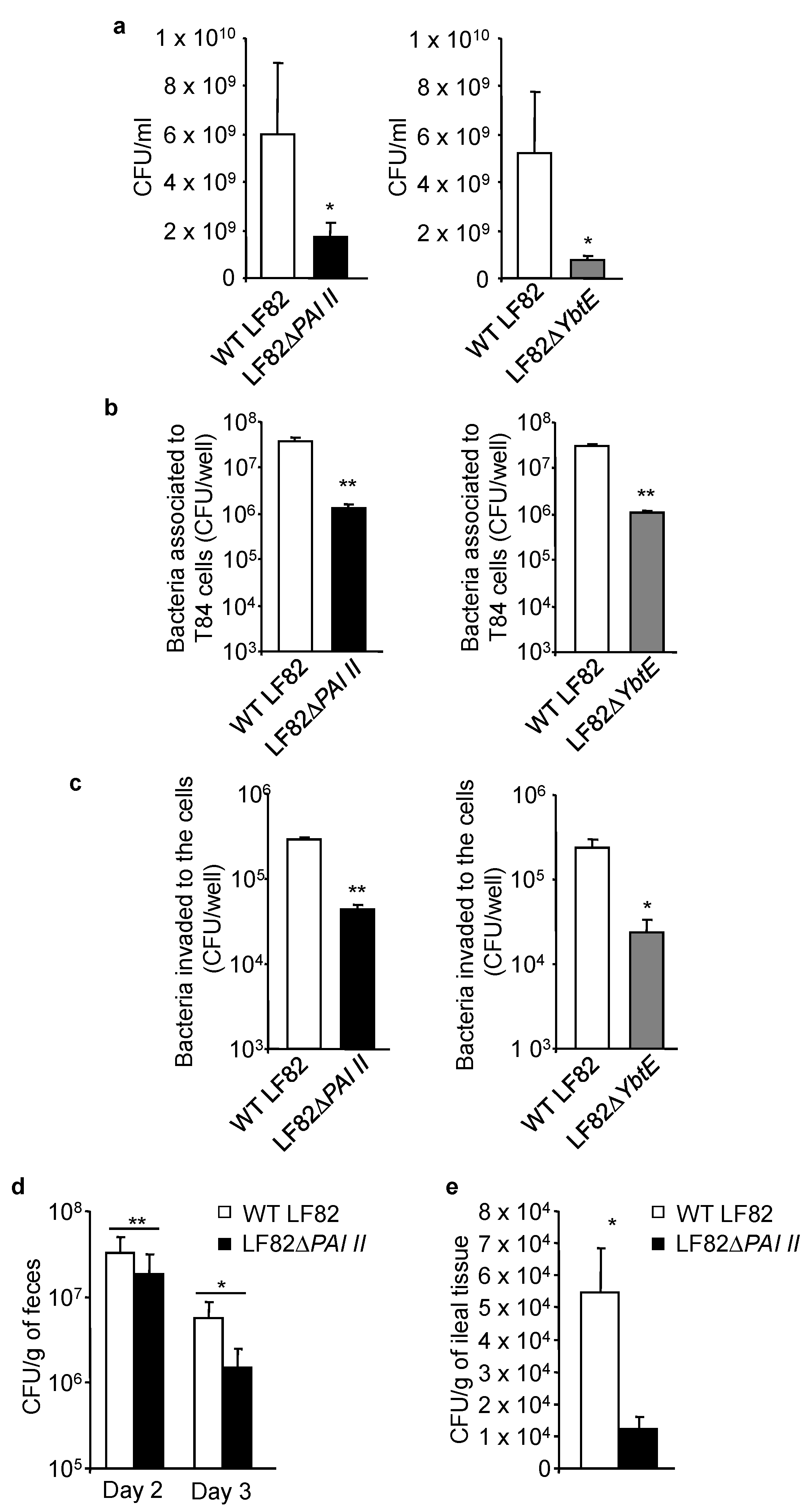
2.3. Loss of Functional Yersiniabactin Impairs AIEC Growth and Colonization of IECs in Competitive Conditions
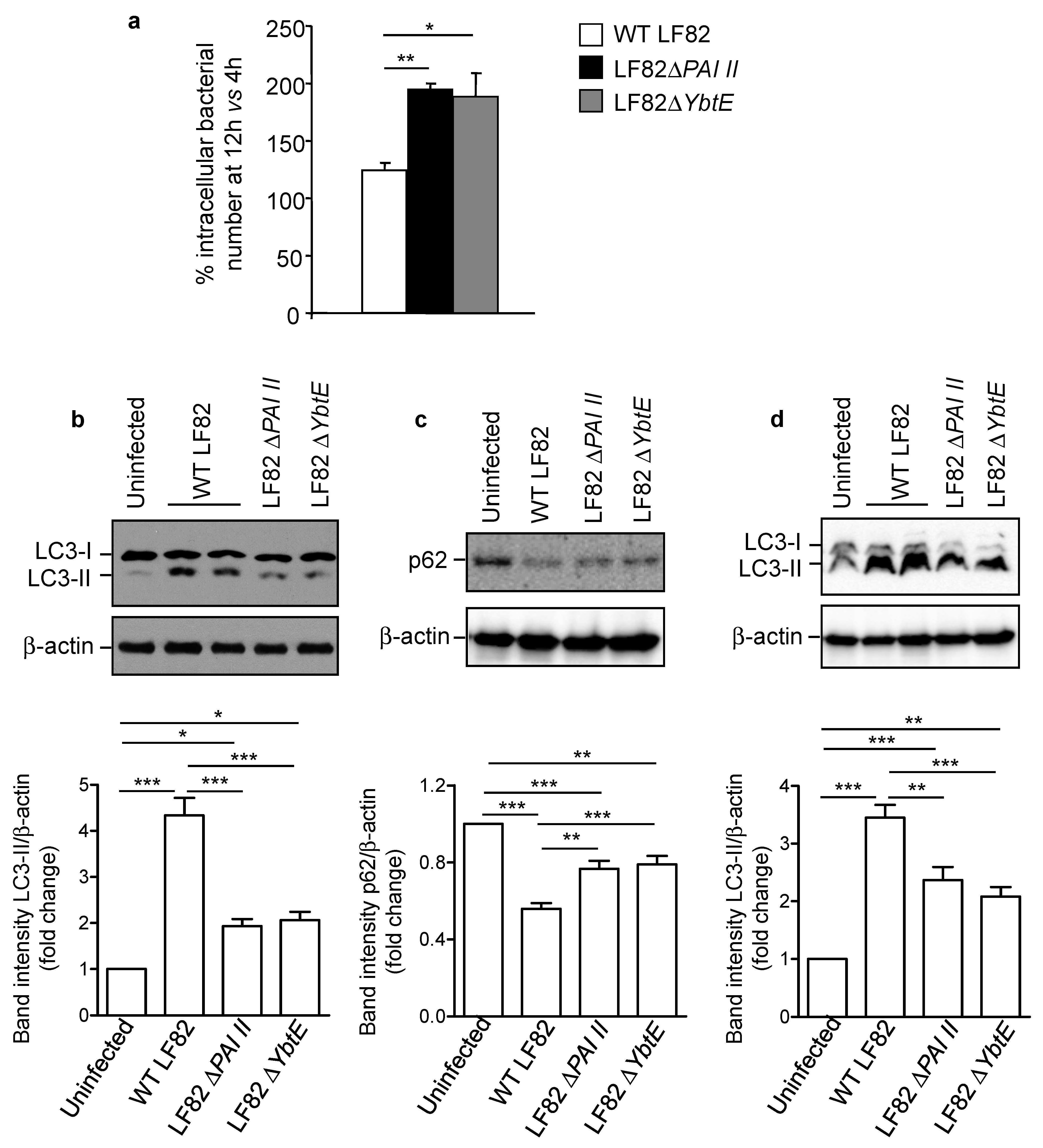
2.4. Loss of Functional Yersiniabactin Increases AIEC Intracellular Proliferation and Decreases Autophagy in IECs
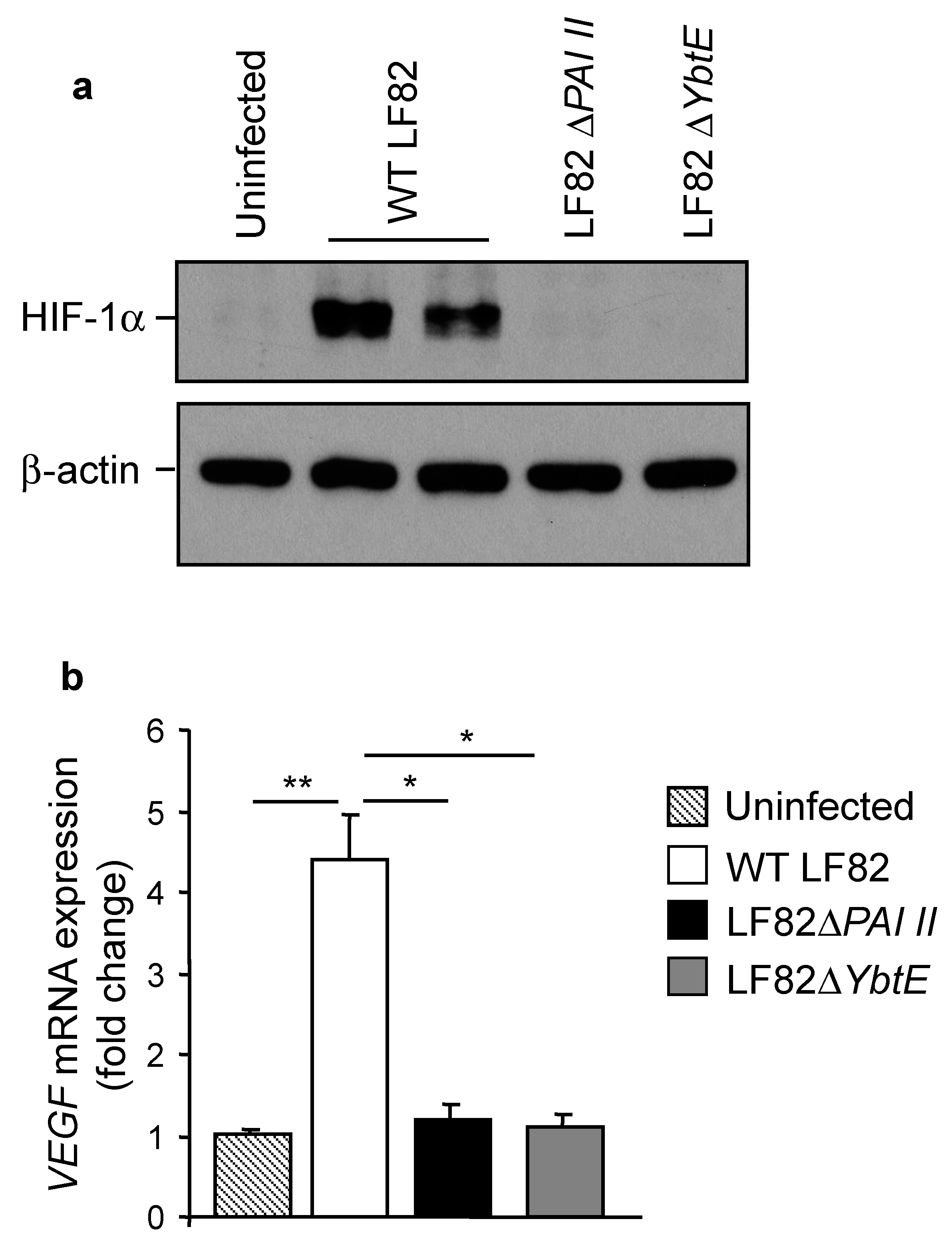
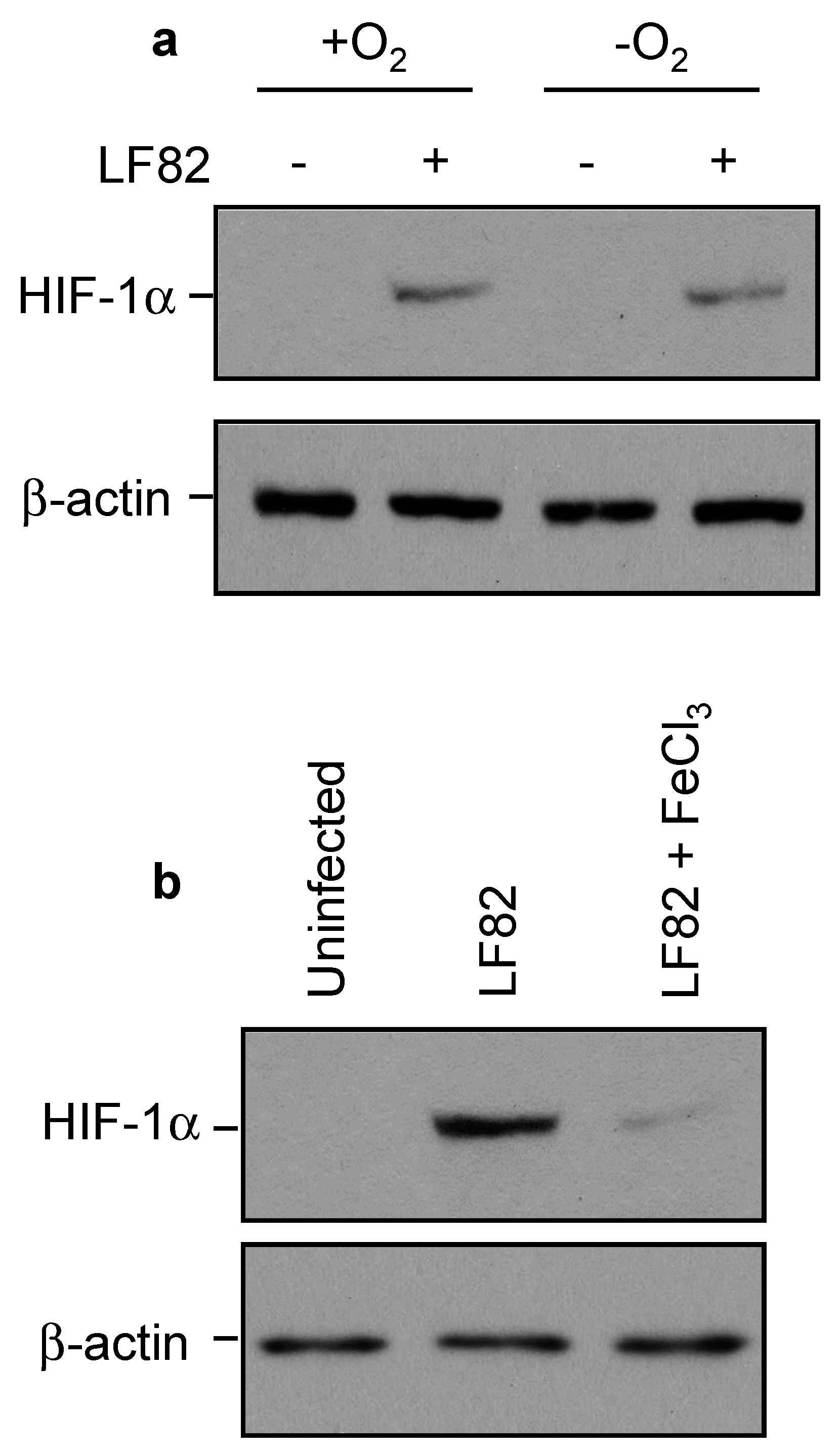
2.5. Loss of Yersiniabactin Inhibits AIEC-Induced HIF-1α Expression and Activation in IECs
2.6. AIEC Induced HIF-1α Expression by Chelating Iron Rather Than by Inducing Hypoxia
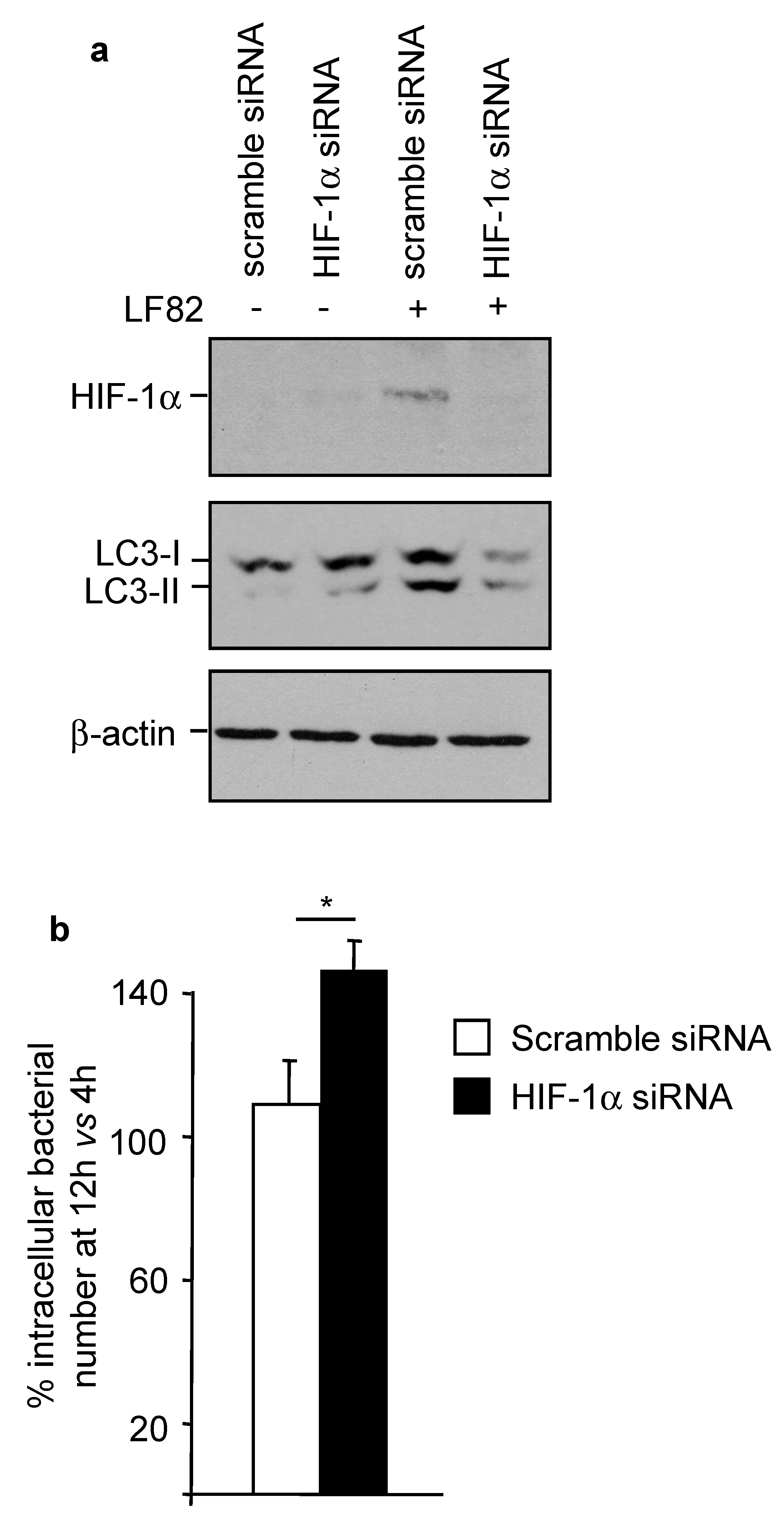
2.7. HIF-1α Controls AIEC Intracellular Replication by Modulating Host Autophagy
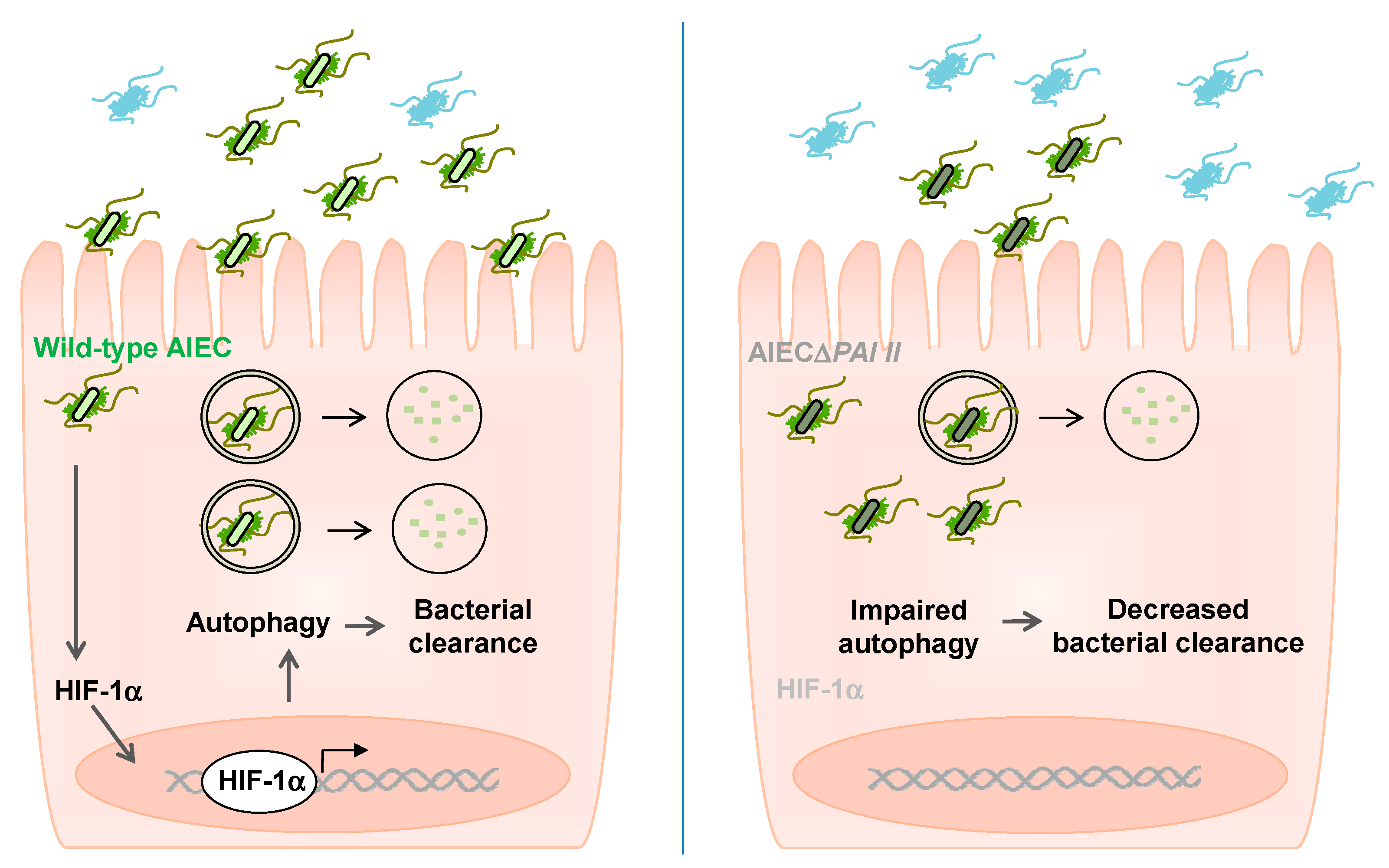
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Screening for the Presence of PAI II by PCR
- LF82_p298Forward: 5′-GCGCCCAGACGAATGTTATT-3′;
- LF82_p298Reverse: 5′-CAACACCTCTCATTACGCGG-3′;
- LF82_p306Forward: 5′-TTCAGGCAGTTTGTGGGAGA-3′;
- LF82_p306Reverse: 5′-TCACTCTTACCGCCAACCAT-3′.
4.3. Construction of AIEC LF82 Chromosomal Mutant Invalidated for PAI I
- LF82_p294Forward: 5′-GGGCCCGAATTCGCTAAGTAATATGCGCCCCG-3′; LF82_p294Reverse: 5′-CGATGTTTCGGAGTCAGTCGCGGATACCTTCACGTTGCTG-3′;
- LF82_p308Forward: 5′-CAGCAACGTGAAGGTATCCGCGACTGACTCCGAAACATCG-3′;
- LF82_p308Reverse: 5′-GGGCCCGGATCCAACGAATTTCCAGGTCGTCG-3′.
- LF82_p298Forward: 5′-GCGCCCAGACGAATGTTATT-3′;
- LF82_p298Reverse: 5′-CAACACCTCTCATTACGCGG-3′;
- LF82_p306Forward: 5′-TTCAGGCAGTTTGTGGGAGA-3′;
- LF82_p306Reverse: 5′-TCACTCTTACCGCCAACCAT-3′.
- YbtEKanaForward: 5′-ATGTGCATCCCGCTGTGGCCCGCCCGGAACGGCAATACTGCGCATCTGGGTAGGCTGGAGCTGCTTCG-3′;
- YbtEKanaReverse: 5′-TCACCTTTCTGAAGTACTGGGCTGTTTTTGCCACGCGGTCATTGCAGCATATGAATATCCTCCTTAGTTC-3′.
4.4. Cell Culture
4.5. Infection and Gentamicin Protection Assay
4.6. Protein Extraction and Western Blot Analysis
4.7. Quantitative Real-Time RT-PCR (qRT-PCR)
- VEGFForward: 5′-TGGAGCGTGTACGTTGGTG-3′;
- VEGFReverse: 5′-GCGAGTCTGTGTTTTTGCAG-3′;
- RPLP0Forward: 5′- TCTGCATTCTCGCTTCCTGG-3′;
- RPLP0Reverse: 5′- CAGGACTCGTTTGTACCCGT-3′.
4.8. Transfection Experiments
4.9. Transmission Electron Microscopy
4.10. Competition in Vitro
4.11. Competition In Vivo
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.-F. Adherent-invasive escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Carrière, J.; Darfeuille-Michaud, A.; Nguyen, H.T.T. Infectious etiopathogenesis of Crohn’s disease. World J. Gastroenterol. 2014, 20, 12102–12117. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.-L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.-F. High prevalence of adherent-invasive escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.L.; Bringer, M.A.; Holt, K.E.; Gordon, D.M.; Dubois, A.L.; Barnich, N.; Darfeuille-Michaud, A.; Pavli, P. Comparative genomics of Crohn’s disease-associated adherent-invasive Escherichia coli. Gut 2017, 66, 1382–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Blanco, J.E.; Blanco, J.; Garcia-Gil, L.J.; Darfeuille-Michaud, A. Molecular Diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Boudeau, J.; Glasser, A.L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasser, A.L.; Boudeau, J.; Barnich, N.; Perruchot, M.H.; Colombel, J.F.; Darfeuille-Michaud, A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chervy, M.; Barnich, N.; Denizot, J. Adherent-invasive E. coli: Update on the lifestyle of a troublemaker in Crohn’s disease. Int. J. Mol. Sci. 2020, 21, 3734. [Google Scholar] [CrossRef]
- Delmas, J.; Gibold, L.; Faïs, T.; Batista, S.; Leremboure, M.; Sinel, C.; Vazeille, E.; Cattoir, V.; Buisson, A.; Barnich, N.; et al. Metabolic Adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci. Rep. 2019, 9, 2175. [Google Scholar] [CrossRef] [PubMed]
- Gibold, L.; Garenaux, E.; Dalmasso, G.; Gallucci, C.; Cia, D.; Mottet-Auselo, B.; Faïs, T.; Darfeuille-Michaud, A.; Nguyen, H.T.T.; Barnich, N.; et al. The vat-AIEC protease promotes crossing of the intestinal mucus layer by Crohn’s disease-associated Escherichia coli. Cell. Microbiol. 2015. [Google Scholar] [CrossRef]
- Sevrin, G.; Massier, S.; Chassaing, B.; Agus, A.; Delmas, J.; Denizot, J.; Billard, E.; Barnich, N. Adaptation of adherent-invasive E. coli to gut environment: Impact on flagellum expression and bacterial colonization ability. Gut Microbes 2020, 11, 364–380. [Google Scholar] [CrossRef]
- Low, D.; Tran, H.T.; Lee, I.A.; Dreux, N.; Kamba, A.; Reinecker, H.C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-binding domains of Escherichia coli chia mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology 2013, 145, 602–612.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudeau, J.; Barnich, N.; Darfeuille-Michaud, A. Type 1 pili-mediated adherence of Escherichia coli strain lf82 isolated from Crohn’s disease is involved in bacterial invasion of intestinal epithelial cells. Mol. Microbiol. 2001, 39, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Dreux, N.; Denizot, J.; Martinez-Medina, M.; Mellmann, A.; Billig, M.; Kisiela, D.; Chattopadhyay, S.; Sokurenko, E.; Neut, C.; Gower-Rousseau, C.; et al. Point mutations in FimH adhesin of Crohn’s disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog 2013, 9, e1003141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnich, N.; Carvalho, F.A.; Glasser, A.L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.F.; et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, F.A.; Barnich, N.; Sivignon, A.; Darcha, C.; Chan, C.H.F.; Stanners, C.P.; Darfeuille-Michaud, A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J. Exp. Med. 2009, 206, 2179–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.T.; Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Autophagy and Crohn’s disease. J. Innate Immun. 2013, 5, 434–443. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell. Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hébuterne, X.; et al. A synonymous variant in IRGM alters a binding site for MiR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Bretin, A.; Carrière, J.; Dalmasso, G.; Bergougnoux, A.; B’chir, W.; Maurin, A.C.; Müller, S.; Seibold, F.; Barnich, N.; Bruhat, A.; et al. Activation of the EIF2AK4-EIF2A/EIF2α-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy 2016, 12, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretin, A.; Lucas, C.; Larabi, A.; Dalmasso, G.; Billard, E.; Barnich, N.; Bonnet, R.; Nguyen, H.T.T. AIEC infection triggers modification of gut microbiota composition in genetically predisposed mice, contributing to intestinal inflammation. Sci. Rep. 2018, 8, 12301. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Dalmasso, G.; Müller, S.; Carrière, J.; Seibold, F.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of micrornas in intestinal epithelial cells to reduce autophagy. Gastroenterology 2014, 146, 508–519. [Google Scholar] [CrossRef]
- Dalmasso, G.; Nguyen, H.T.T.; Faïs, T.; Massier, S.; Barnich, N.; Delmas, J.; Bonnet, R. Crohn’s disease-associated adherent-invasive Escherichia coli manipulate host autophagy by impairing SUMOylation. Cells 2019, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Carrière, J.; Bretin, A.; Darfeuille-Michaud, A.; Barnich, N.; Nguyen, H.T.T. Exosomes released from cells infected with Crohn’s disease-associated adherent-invasive Escherichia coli activate host innate immune responses and enhance bacterial intracellular replication. Inflamm. Bowel Dis. 2016, 22, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Larabi, A.; Dalmasso, G.; Delmas, J.; Barnich, N.; Nguyen, H.T.T. Exosomes transfer MiRNAs from cell-to-cell to inhibit autophagy during infection with Crohn’s disease-associated adherent-invasive E. coli. Gut Microbes 2020, 11, 1677–1694. [Google Scholar] [CrossRef] [PubMed]
- Evstatiev, R.; Gasche, C. Iron sensing and signalling. Gut 2012, 61, 933–952. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.R.; Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Siderophores in iron metabolism: From mechanism to therapy potential. Trends Mol. Med. 2016, 22, 1077–1090. [Google Scholar] [CrossRef] [Green Version]
- Ellermann, M.; Arthur, J.C. Siderophore-mediated iron acquisition and modulation of host-bacterial interactions. Free Radic. Biol. Med. 2017, 105, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, Nature and utility of universal iron chelator-siderophore: A review. Microbiol. Res. 2017, 212–213, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Rakin, A.; Heesemann, J. The yersinia high-pathogenicity island (HPI): Evolutionary and functional aspects. Int. J. Med. Microbiol. 2004, 294, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Peyretaillade, E.; Claret, L.; de Vallée, A.; Dossat, C.; Vacherie, B.; Zineb, E.H.; Segurens, B.; Barbe, V.; Sauvanet, P.; et al. Complete genome sequence of Crohn’s disease-associated adherent-invasive E. coli strain LF82. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.K.; Kamm, M.A.; Teo, S.M.; Inouye, M.; Wagner, J.; Kirkwood, C.D. Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: A systematic review. Inflamm. Bowel Dis. 2015, 21, 1219–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faber, F.; Bäumler, A.J. The impact of intestinal inflammation on the nutritional environment of the gut microbiota. Immunol. Lett. 2014, 162, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Céspedes, S.; Saitz, W.; Del Canto, F.; De la Fuente, M.; Quera, R.; Hermoso, M.; Muñoz, R.; Ginard, D.; Khorrami, S.; Girón, J.; et al. Genetic diversity and virulence determinants of Escherichia coli strains isolated from patients with Crohn’s disease in Spain and Chile. Front. Microbiol. 2017, 8, 639. [Google Scholar] [CrossRef] [PubMed]
- Rakitina, D.V.; Manolov, A.I.; Kanygina, A.V.; Garushyants, S.K.; Baikova, J.P.; Alexeev, D.G.; Ladygina, V.G.; Kostryukova, E.S.; Larin, A.K.; Semashko, T.A.; et al. Genome analysis of E. coli isolated from Crohn’s disease patients. BMC Genomics 2017, 18, 544. [Google Scholar] [CrossRef] [Green Version]
- Dogan, B.; Suzuki, H.; Herlekar, D.; Sartor, R.B.; Campbell, B.J.; Roberts, C.L.; Stewart, K.; Scherl, E.J.; Araz, Y.; Bitar, P.P.; et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm. Bowel Dis. 2014, 20, 1919–1932. [Google Scholar] [CrossRef]
- Bearden, S.W.; Fetherston, J.D.; Perry, R.D. Genetic organization of the yersiniabactin biosynthetic region and construction of avirulent mutants in yersinia pestis. Infect. Immun. 1997, 65, 1659–1668. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 8, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.; Eltzschig, H.K.; Wurz, H.; Hantke, K.; Rakin, A.; Yazdi, A.S.; Matteoli, G.; Bohn, E.; Autenrieth, I.B.; Karhausen, J.; et al. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 2008, 134, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of Vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1, O2, and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Hartmann, F.; Dignass, A.U. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Bachman, M.A.; Oyler, J.E.; Burns, S.H.; Caza, M.; Lépine, F.; Dozois, C.M.; Weiser, J.N. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect. Immun. 2011, 79, 3309–3316. [Google Scholar] [CrossRef] [Green Version]
- Heesemann, J.; Hantke, K.; Vocke, T.; Saken, E.; Rakin, A.; Stojiljkovic, I.; Berner, R. Virulence of Yersinia enterocolitica is closely associated with siderophore production, expression of an iron-repressible outer membrane polypeptide of 65,000 Da and pesticin sensitivity. Mol. Microbiol. 1993, 8, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ellermann, M.; Gharaibeh, R.Z.; Fulbright, L.; Dogan, B.; Moore, L.N.; Broberg, C.A.; Lopez, L.R.; Rothemich, A.M.; Herzog, J.W.; Rogala, A.; et al. Yersiniabactin-Producing adherent/invasive Escherichia coli promotes inflammation-associated fibrosis in gnotobiotic Il10-/- mice. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massier, S.; Miquel, S.; Dreux, N.; Agus, A.; Bouhours, S.; Denizot, J.; Darfeuille-Michaud, A.; Barnich, N. Involvement of type VI secretion systems in virulence of adherent-invasive Escherichia coli isolated from patients with Crohn’s disease. J. Crohns Colitis 2015, 9, S67–S68. [Google Scholar] [CrossRef]
- Pósfai, G.; Kolisnychenko, V.; Bereczki, Z.; Blattner, F.R. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999, 27, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaveroche, M.K.; Ghigo, J.M.; d’Enfert, C. A Rapid method for efficient gene replacement in the filamentous fungus aspergillus nidulans. Nucleic Acids Res. 2000, 28, e97. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalmasso, G.; Nguyen, H.T.T.; Faïs, T.; Massier, S.; Chevarin, C.; Vazeille, E.; Barnich, N.; Delmas, J.; Bonnet, R. Yersiniabactin Siderophore of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Is Involved in Autophagy Activation in Host Cells. Int. J. Mol. Sci. 2021, 22, 3512. https://doi.org/10.3390/ijms22073512
Dalmasso G, Nguyen HTT, Faïs T, Massier S, Chevarin C, Vazeille E, Barnich N, Delmas J, Bonnet R. Yersiniabactin Siderophore of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Is Involved in Autophagy Activation in Host Cells. International Journal of Molecular Sciences. 2021; 22(7):3512. https://doi.org/10.3390/ijms22073512
Chicago/Turabian StyleDalmasso, Guillaume, Hang Thi Thu Nguyen, Tiphanie Faïs, Sébastien Massier, Caroline Chevarin, Emilie Vazeille, Nicolas Barnich, Julien Delmas, and Richard Bonnet. 2021. "Yersiniabactin Siderophore of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Is Involved in Autophagy Activation in Host Cells" International Journal of Molecular Sciences 22, no. 7: 3512. https://doi.org/10.3390/ijms22073512