1. Introduction
Retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA) are inherited retinal degenerative diseases causing progressive vision loss, ultimately leading to blindness. Mutations in the Crumbs homolog 1 (
CRB1) gene is a frequent cause of these retinal dystrophies in humans [
1]. The
CRB1 gene, mapped to chromosome 1q31.3, encodes a large transmembrane protein and belongs to the Crumbs (CRB) family, with family members CRB2 and CRB3. CRB proteins are located in the subapical region above adherens junctions at the outer limiting membrane (OLM), where it can interact with, amongst others, PALS1 to form the canonical Crumbs complex [
2,
3,
4].
Crb1 knockout (
Crb1KO) mouse models show a mild retinal degeneration with OLM disruptions and ectopic rows of photoreceptor cell nuclei in the photoreceptor segment layers from postnatal day 14 (P14) [
5]. Concomitant loss of
Crb2 in
Crb1KO mice results in a more severe RP or LCA phenotype, depending on which cell type lacks
Crb2 [
6,
7,
8,
9]. Currently, there is no treatment available for
CRB1-related retinal dystrophies.
A Brown Norway rat strain was described with an inherited retinal degenerative phenotype caused by a spontaneous in frame insertion-deletion (indel) in exon 6 of the
Crb1 gene [
10]. These
Crb1 mutant rats, expressing an alternative CRB1
INDEL protein, exhibit an early-onset loss of retinal function from 3 weeks of age. In addition, the first signs of retinal degeneration were observed from P15, including OLM disruptions and ectopic rows of photoreceptor cell nuclei in the photoreceptor segment layers. At older ages, these disruptions progress and ultimately lead to a focal loss of retinal lamination [
10]. Because of its naturally occurring mutation and early-onset severe retinal phenotype, these rats are a potential attractive animal model for the development of gene therapy. Currently, gene therapy using adeno-associated viral vectors (AAVs) is the leading platform of gene delivery for the treatment of retinal dystrophies because of its low toxicity and ability to target both dividing and non-dividing cells [
11]. In addition, different AAV capsids display distinct cell tropisms, making it possible to target different cell types. Moreover, previous mouse gene supplementation studies with AAV expressing human
CRB2 (h
CRB2) have shown to preserve the retinal morphology and function in
Crb1 RP mouse models [
12,
13]. Altogether, this indicates the potential use of AAV-mediated gene therapy for
CRB1-related retinal dystrophies.
Here, we perform a thorough characterization of the morphological phenotype of new-born and adult Crb1 mutant rats and its effect on retinal function and visual acuity in comparison with age-matched control rats. Using immuno-electron-microscopy, we show that, in control rats, canonical CRB1 localizes specifically in Müller glial cells (MGCs) at the subapical region adjacent to the adherens junctions at the outer limiting membrane (OLM), whereas the Crb1 mutant rats barely express detectable levels of CRB1INDEL at the subapical region of MGCs. In turn, CRB2 is localized at the subapical region in MGCs and photoreceptors of both control and Crb1 mutant rats. Next, we describe the tropism of three different AAV serotypes (AAV5-, AAV9- and AAV6-variant ShH10Y445F) expressing GFP driven by the cytomegalovirus (CMV) promoter in new-born control and Crb1 mutant rat retinas. Tropism data at P5 and P8 shows that subretinal injection of AAV5.CMV.GFP and AAV9.CMV.GFP mainly transduces photoreceptors and retinal pigment epithelium (RPE), whereas intravitreal injection at P5 or P8 of ShH10Y445F.CMV.GFP efficiently transduces MGCs. Based on this knowledge, AAV-mediated hCRB1 and hCRB2 gene therapy were explored in P5 Crb1 mutant rats.
3. Discussion
In this study, we demonstrate (1) progressive retinal degeneration in Crb1 mutant rats, causing loss of visual and retinal function; (2) ultrastructural localization of CRB1 at the subapical region of MGCs, and CRB2 at the subapical region of MGCs and photoreceptors in control rats; (3) a decrease in CRB1INDEL at the subapical region of MGCs in Crb1 mutant rats; (4) tropism by subretinal and intravitreal application of AAV5, AAV9 and ShH10Y in control and Crb1 mutant rats; and (5) AAV-hCRB gene therapy at P5 for MGCs of Crb1 mutant rats does not result in a functional rescue.
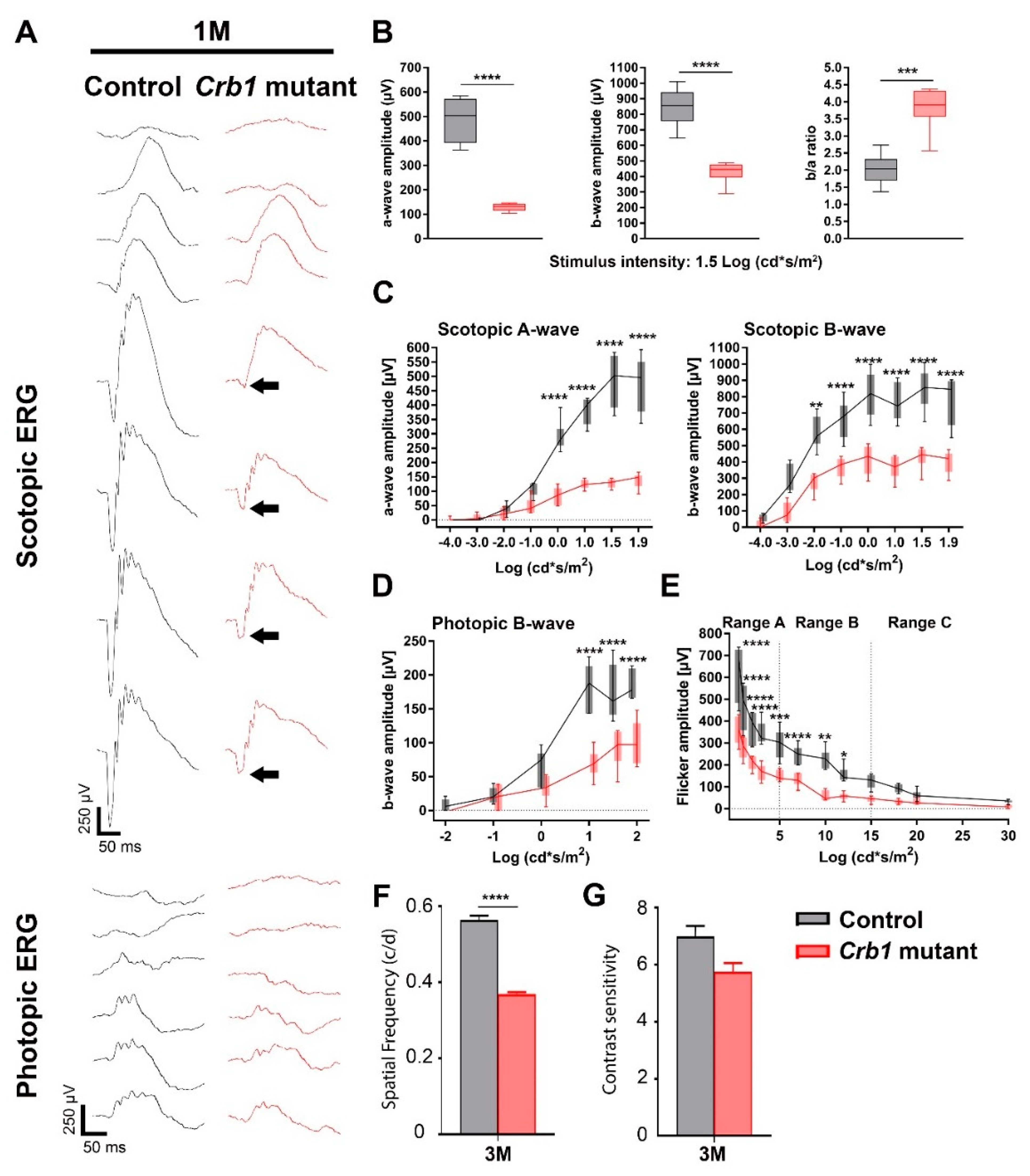
The
Crb1 mutant rats exhibit a significant decreased ERG response at 1 month of age in comparison with age-matched control rats, which is in accordance with previously published results comparing the
Crb1 mutant to BN-Harlan rats [
10]. We further expand the characterization of the
Crb1 mutant rat strain by showing a reduced OKT spatial frequency response in 3-month-old
Crb1 mutant rats compared to age-matched control rats. Interestingly, although the control rats show a reduced ERG response from 1 month of age onwards, the OKT spatial frequency decline is minor. This discrepancy has been described before, where similar spatial frequencies only declined months after significant photoreceptor and ERG response loss [
20].
Here, we observed the first signs of retinal degeneration from P10 in the
Crb1 mutant rat retina, including photoreceptor nuclei protrusions in the photoreceptor segment layers and OLM breaks at foci throughout the entire retina. These disruptions are ultimately resulting in large regions with a complete disorganized ONL and INL lamination in adult
Crb1 mutant rats. Interestingly, the phenotype at P10 in the
Crb1 mutant rats is comparable to previously described
Crb1 mouse models [
3,
17], but the retinal phenotype in these rats at 1 month of age is much more severe. In addition, in
Crb1 mouse models, the retinal degeneration is limited to the inferior quadrant [
3,
17], whereas in the
Crb1 mutant rats it is presented throughout the entire retina. These discrepancies could be because of (1) different genetic backgrounds, including species differences; (2) different types of mutations affecting different CRB isoforms [
21,
22], thereby expressing a distinct CRB1
INDEL protein in the
Crb1 mutant rats; or (3) the total expression levels of CRB2 might be significantly lower in new-born rats compared to new-born mice. Decreased levels of CRB2 or dysregulation of other CRB-interacting proteins could result in less stabilization of the adherens junction complex at the OLM, resulting in a more severe phenotype. In addition, transcriptomic analysis identified several dysregulated pathways in
Crb1 mutant rats, such as TGF-β signaling, matrix metalloproteinases, type II interferon signaling, MAPK cascade, inflammatory pathways, regulation of actin cytoskeleton and many more [
10]. More research is required to define which factors play a major role in the retinal degeneration in these
Crb1 mutant rats.
SD-OCT imaging allows to follow the retinal degeneration over time in
Crb1 mutant compared to age-matched control rat retinas. Previous research using SD-OCT with
Crb1 rd8 or
Crb1lowMGC mouse models show typical ocular lesions, such as pseudo-rosette formation [
13,
23,
24]. Here, we observed a relatively healthy retinal lamination in P17
Crb1 mutant rat retina with some sporadic disruptions at the OLM. At one month of age, the retinal lamination is disrupted throughout the retina, indicated by hyperreflective regions at the ONL. These hyperreflective disruptions are thought to begin in the OLM because of adhesion abnormalities between the photoreceptor and MGCs. Over time, in 2- and 3-month-old rats, an increased degeneration with larger hyperreflective regions, indicating a potential lack of retinal lamination, is observed. Similar results were obtained using histological sections. Like human
CRB1-RD patients [
24], hyperreflective lesions of various sizes were observed in the
Crb1 mutant rat starting at 1 month of age. The hyperreflective ONL and INL in the
Crb1 mutant rats made automated as well as manual segmentation of the layers challenging, particularly at 2 and 3 months of age. Therefore, we quantified the length of the observed disruptions and compared it to the length of the retinal view, hereby distinguishing between the length of disrupted and normal laminated retinas. Quantification of the data showed a significant increase in OLM breaks and disruptions at the OPL in the
Crb1 mutant compared to age-matched control rats at 1, 2 and 3 months of age. Several researchers have shown that quantification of retinal layer size by SD-OCT, compared with histological sections, contain less variations, presumably due to artefacts from the post-mortem processing [
25]. Altogether, these data are showing the importance of the SD-OCT imaging over time in
Crb1 mutant rats.
Immunohistochemical analysis reveal the localization of CRB2 at the subapical region (SAR) in control and
Crb1 mutant rats, while canonical CRB1 is only detected at the SAR of control rats. The CRB1 antibody used detects the carboxyl terminus of the CRB1 protein, and our data show that the
Crb1 mutant rats lack the full length CRB1 at the OLM. In control rats, subcellular localization of CRB1 and CRB2 by immuno-electron microscopy revealed the presence of CRB1 at the SAR adjacent to the adherens junctions in MGCs, and of CRB2 at the SAR in both MGCs and photoreceptors. These data correspond well to the localization previously found in mouse studies [
26]. In postmortem human cadaver retinas, however, CRB1 localized at the subapical region in both MGCs and photoreceptor. In these studies, CRB2 localized at the SAR in MGCs, with CRB2 localized at vesicles in the photoreceptor inner segments [
12,
27]. Interestingly, recent data revealed the subcellular localization of both CRB1 and CRB2 at the SAR of MGCs as well as photoreceptors in human iPSC-derived retinal organoids, second trimester human fetal retina and non-human-primate retinas [
8,
19]. The cellular localization of CRB1 and CRB2 in Brown Norway rats are therefore only similar to mice.
The
Crb1 gene locus is complex with various
Crb1 splice forms and gene products from alternate promoters, such as
Crb1-B [
17,
21,
28]. Full-length canonical
Crb1 is encoded on 12 exons with the highly conserved CRB1 carboxyl terminus encoded on exon-12, whereas the
Crb1-B lacks the conserved carboxyl terminus since encoded on 5′-alternate exon-5a, 6–11 alternate-3′ [
21]. The in-frame insertion-deletion mutation in exon 6 of the
Crb1 mutant rat also affects the
Crb1-B gene, and it is to be tested whether the mutation specifically in
Crb1-B contributes to the observed early-onset severe retinal phenotype. Interestingly,
Crb1 rd8 mice have an out-of-frame base-pair deletion in exon-9 that causes an alternate-3′ and encodes, therefore, for two truncated transcripts (upstream promoter driving expression of
Crb1 exons 1–6 indel, 7–9 alternate-3′; and a promoter in intron-5 driving expression of
Crb1 exons 5a-6indel, 7–9 alternate-3′) [
17]. However, the mutation affecting these two gene products from
Crb1 rd8 do not result in a severe retinal degeneration as observed in the
Crb1 mutant rats, suggesting the existence of other modifying factors that play a role in the severity of the retinal phenotype. Reduced levels of CRB2 in MGCs and/or photoreceptors or retinal progenitors in mice lacking CRB1 result in a significant more severe retinitis pigmentosa or Leber congenital amaurosis retinal phenotype [
8,
27]. Altogether, these data suggest the existence of other modifying factors that play a role in the severity of the retinal phenotype observed in
Crb1 mutant rats. It remains of interest to test whether low levels of CRB2 contribute to the early-onset severe retinal phenotype observed in new-born
Crb1 mutant rats, whereas high levels of CRB2 might suppress the onset of retinal phenotype in
Crb1 mice.
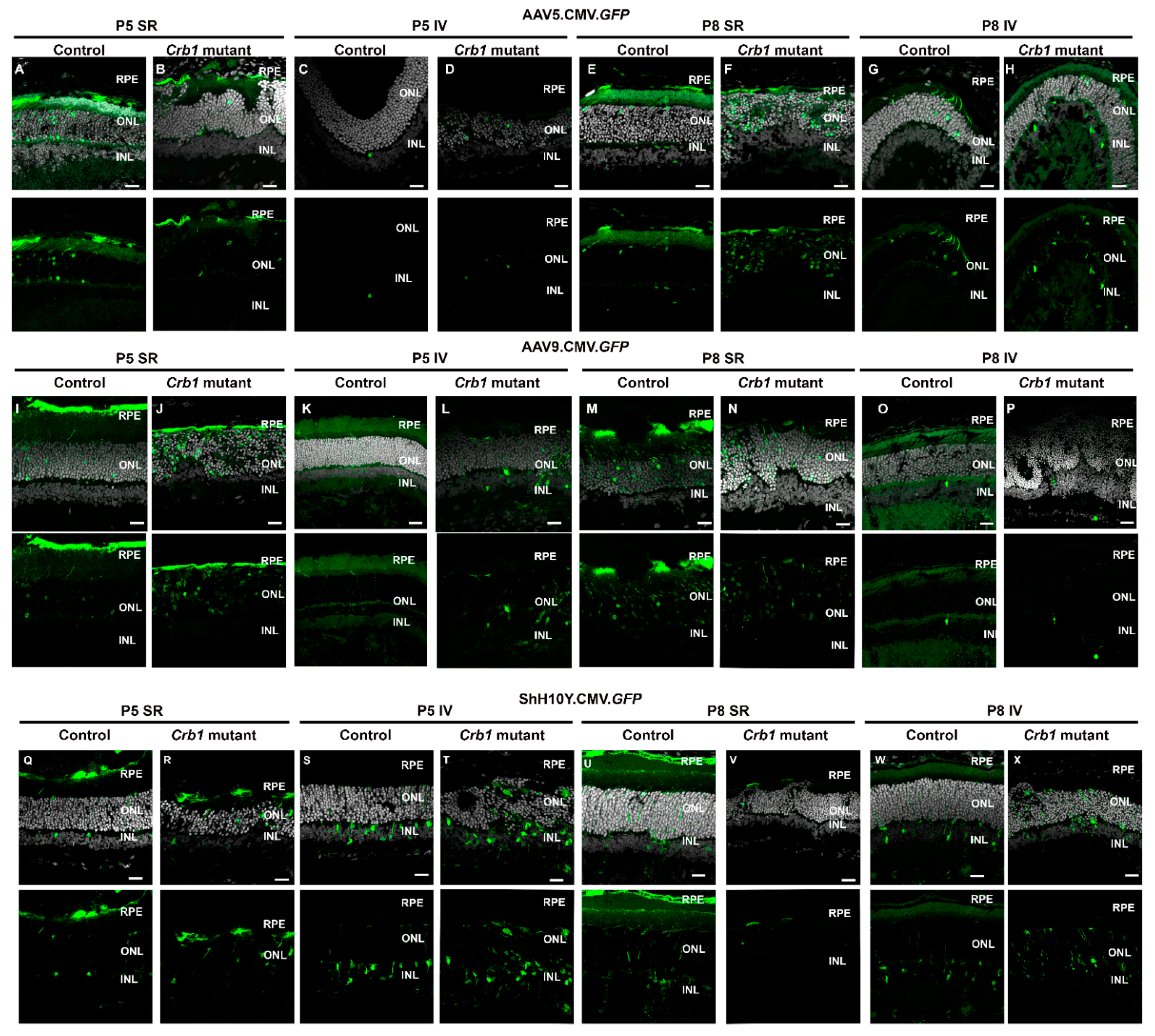
For AAV-mediated gene therapy purposes, in new-born
Crb1 mutant rats, we determined the retinal tropism of three different AAV serotypes (AAV5, AAV9, ShH10Y) upon different routes of AAV delivery. The AAV capsids were injected at different time points (P5 or P8), either subretinal or intravitreal, in both control and
Crb1 mutant rats. Even though retinal injection efficiency can vary between animals, the transduced layers of cells with a specific capsid and type of injection was similar between animals injected at the two different time points. With subretinal delivery, the serotypes AAV5 and AAV9 successfully transduced the RPE and photoreceptors in new-born control and
Crb1 mutant rats, while subretinal delivery of ShH10Y was transducing RPE, photoreceptors, MGCs and other cell types in the INL. Subretinal delivery of AAV9 at P5 and P8 in both control and
Crb1 mutant rats results in a similar transduction pattern as observed by others after subretinal injection in two-month old Sprague-Dawley rats [
29]. In addition, AAV5 tropism at P5 and P8 in control and
Crb1 mutant rats is similar as described in 6- to 8-week-old C57/BL6 wild-type mice [
30]. When AAV.CMV.
GFP vectors were injected intravitreally in new-born control and
Crb1 mutant rats, serotypes AAV5 and AAV9 showed a relatively poor transduction efficiency of both photoreceptors and other cells in the INL. Other researchers showed a poor transduction of cells in the INL as well upon intravitreal delivery of AAV2, AAV6 and AAV8 in two-month-old Sprague-Dawley rats [
29]. However, we observed efficient transduction of mainly MGCs in the INL after intravitreal injection in new-born control and
Crb1 mutant rats with ShH10Y, which is consistent with previously published data obtained in the adult rat [
31].
Transduction efficiency in the degenerated retina might differ from that in the healthy control retina [
30,
32]. However, we did not observe a difference in cellular tropism nor in the transduction efficiency between control and
Crb1 mutant rats with all serotypes and the expression vector AAV.CMV.
GFP tested. Moreover, no differences in tropism were observed when all AAV serotypes were injected either at P5, before the onset of retinal degeneration or at P8, closer to the first signs of degeneration. This might be explained by the correctly laminated retina in the new-born
Crb1 mutant rat retina, and it is well possible that AAV tropism might differ when injected at later time points where the degeneration is more advanced. In summary, we highlight the important differences of AAV capsids and types of delivery that could be considered with future gene therapy approaches.
Finally, we explored the possibility of AAV-mediated h
CRB gene therapy in
Crb1 mutant new-born rats. Based on the tropism results, we used intravitreal injection of ShH10Y-h
CRB1 and ShH10Y-h
CRB2 at P5 in
Crb1 mutant rats for our gene therapy experiments. The efficacy of these ShH10Y-h
CRB gene therapy vectors was shown previously [
12,
13]. When individual untreated
Crb1 mutant rat ERG responses were analyzed at different time points, no differences were detected between the left and right eyes within the same animal. This suggests that both eyes show similar rates of loss of retinal function.
Lack of an improved visual function after AAV-
CRB application could be caused by the time lag for AAV-mediated gene therapy; it might take several days to weeks before the h
CRB transgene in
Crb1 mutant rats is expressed at full level in the target cells, in this case the target cells being the MGCs. In our tropism studies, we observed upon intravitreal injection of 1 µL 1 × 10
13 gc/mL ShH10Y-h
CRB at P5 a good but potentially incomplete transduction of
Crb1 mutant rat MGCs. In other studies, an increased b-wave was observed when 10-day-old Royal College of Surgeons rats were subretinally injected with a total of 8 µL containing 4 × 10
8 particles (4 µL superior hemisphere and 4 µL in the inferior hemisphere) of AAV2.CMV.
Mertk to target defective RPE [
33]. Whereas, in our
Crb1 mutant rat studies, 1 µL of a dose of 1 × 10
13 gc/mL was injected intravitreally at P5, which might be a too low dose of ShH10Y-h
CRB1 or ShH10Y-h
CRB2 to slow down the retinal degeneration observed in the
Crb1 mutant rats. In addition, 1 µL of our gene therapy vector might not spread well throughout the entire retina. Improving the surgical application technique to enlarge the transduced area of the retina could be considered [
34]. Moreover, we observed a wide variability in ERG response between different
Crb1 mutant rat litters, likely because of the Brown Norway genetic background. Backcrossing these rats into a more defined genetic background might decrease the observed variability in retinal phenotype. In
Crb1rd8/rd8 mice on a C57BL/6 genetic background, considerable variation in retinal phenotype was observed as well [
35]. In addition, the injection efficiency and thereby the number of AAV viral particles taken up by the retinal cells might vary between animals. Moreover, there might be differences in intracellular release of AAV-DNA from capsids inside the targeted cells. Finally, proof-of-concept studies showed functional and structural preservation in a
Crb1 mouse model by using AAV2/9.CMV.h
CRB2 [
12] or AA2/ShH10Y.CMV.h
CRB2 [
13]. However, the retinal phenotypes in these mice were less severe than the one observed at 1 and 3 months of age
Crb1 mutant rats. In previous studies, we showed that human CRB2 can compensate for the loss of endogenous Crb proteins in the mouse retina [
12,
13]. Here, we tested human
CRB1 or
CRB2 in gene therapy studies to compensate for the loss of functional Crb1 protein in the rat retina. We showed expression of h
CRB1 in
Crb1 mutant rats after intravitreally injected ShH10Y-h
CRB1 (
Figure S3C), but we did not observe an enhanced retinal function after gene therapy delivery. In silico analysis show that both CRB1 and CRB2 are highly conserved proteins between rat and human (
Figure S4). Despite the high sequence similarity, proteins may have distinct species-specific properties or activity that need to be taken into account when designing gene therapy studies. Here, we did not test the application of rat
Crb1 or
Crb2 gene therapy vectors to the rat retina due to ethical issues, as the expected outcome of such experiments on a large group of rats is presumed to be negative. Therefore, we cannot exclude the possibility that rat Crb1 or Crb2 proteins could alleviate the loss of endogenous rat Crb1 in models with slower onset of retinal degeneration. All these differences could explain the lack of functional rescue observed here; thus, future experiments could focus on the development of novel gene therapy vectors that allow immediate early-onset expression of transgenes at P5 or low molecular weight drug therapy in new-born rats, or in utero gene therapy approaches.
In conclusion, we further characterized the early-onset morphological phenotype and the retinal function of Crb1 mutant rats in comparison to age-matched control rats. In addition, we show that the ultrastructural localization of endogenous CRB1 and CRB2 in the rat retina is similar as observed in the mouse retina. In addition, as little is known about AAV tropism administered in new-born Brown Norway rats, we showed differences in cellular tropism when three different AAV capsids were injected in both Crb1 mutant and control rats. Finally, no timely rescue of the retinal phenotype was observed using retinal function and visual acuity, suggesting the need for earlier onset of expression of recombinant hCRB proteins in Müller glial cells to rescue the severe retinal phenotype in Crb1 mutant rats.
4. Materials and Methods
4.1. Animals
Procedures concerning animals were performed in accordance with the EU Directive 2010/63/EU for animal experiments and with permission of the Dutch Central Authority for Scientific Procedures on Animals (CCD), permit number 1160020172924, approved 18, January, 2018. The animals were maintained on a 12 h day–night cycle and were supplied ad libitum with food and water. Brown Norway rats from Janvier Labs with a spontaneous mutation in the
Crb1 gene were used in this study [
10]; the
Crb1 mutant rat breeding was set up within the LUMC animal facility. Age-matched control Brown Norway rats lacking the mutation in the
Crb1 gene from Charles River Laboratories were used as controls. Animals were killed by carbon dioxide inhalation.
4.2. DNA Isolation and Genetic Analysis
The presence of spontaneous in-frame insertion-deletion (indel) in exon 6 of the Crb1 gene was validated using DNA extraction from rat’s tails by proteinase K digestion overnight at 55 °C in lysis buffer (100 mM Tris-HCl, pH 8.5, 5 mM EDTA, 0.2% SDS, and 200 mM NaCl). Biopsies were centrifuged for 15 min, the supernatant was transferred and mixed vigorously with isopropanol, followed by a 10 min centrifuge at 14,000 rpm and removal of the supernatant. Then, the pellet was washed twice with 80% ethanol, air dried for 10 min at 55 °C and subsequently resuspended in 200 μL 10% TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). Finally, DNase was inactivated for 15 min at 65 °C. For genotyping, PCR with primers targeting the location of the INDEL in the Crb1 gene was performed and was subsequently sequenced using Sanger Sequencing, FW: 5′-TTCAGACTGTTCAGCCAAATGC-3′, REV: 5′-TGTCCCCATTGGTAAGCCACC-3′.
4.3. Electroretinography (ERG)
Dark- and light-adapted ERGs were performed under dim red light using an Espion E2 (Diagnosys, LLC, MA). ERGs were performed on 1, 2 and 3-month-old control and
Crb1 mutant rats. Rats were anesthetized using 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneally and the pupils were diluted using tropicamide drops (5 mg/mL). Rats were placed on a heating pad, and reference and ground platinum electrodes were placed subcutaneously and in the base of the tail, respectively. ERGs were recorded from both eyes using gold wire electrodes. Hypromellose eye drops (3 mg/mL, Teva) were given between recordings to prevent eyes from drying. Single (scotopic and photopic ERG) or brief train (Flicker ERG) white (6500k) flashes were used. The band-pass filter frequencies were 0.3 and 300 Hz. Scotopic recordings were obtained from dark-adapted animals at the following light intensities: -4, -3, -2, -1, 0, 1, 1.5 and 1.9 log cd s/m2 [
36]. Flicker recordings were obtained under a fixed light intensity of 0.5 log cd s/m2 with varying frequency (0.5, 1, 2, 3, 5, 7, 10, 12, 15, 18, 20 and 30 Hz) [
37,
38]. Photopic recordings were performed following 10 min light adaptation on a background light intensity of 30 cd*m2 and the light intensity series used was: -2, -1, 0, 1, 1.5, 1.9 log cd*s/m2 [
36]. The numbers of rats used per time point are indicated under the designated figure. For the gene therapy studies, responses of the treated eye, either right or left eye, were compared with the other eye, mock injected (PBS), at each time point analyzed.
4.4. Optokinetic Tracking Reflex (OKT)
Spatial frequency and contrast sensitivity thresholds were measured using an optomotor system (Cerebral Mechanics, Lethbridge, AB, Canada). Two and three month(s)-old rats were placed on a small platform in the center of four computer monitors that formed a virtual drum with a rotating vertical sine wave grating (12°/s (d/s)), as described previously [
14]. Head movements in the same direction as the rotating gratings were considered as positive responses and no response was considered as negative response. Spatial frequency thresholds were determined with an increasing staircase paradigm, starting at 0.042 cycles/deg (c/d) with 100% contrast. Contrast sensitivity thresholds were measured across three spatial frequencies (0.092 c/d). The reciprocal of the contrast sensitivity threshold was used as the contrast sensitivity value at each spatial frequency.
4.5. Morphological Analysis
Eyes were collected at a range of time points from P5 to 3-month-old control and
Crb1 mutant rats (
n = 2–4/age/group). For morphological analysis, eyes were enucleated and fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 20 min at room temperature. After fixation, the eyes were dehydrated for 30 min in 30, 50, 70, 90 and 99% ethanol. Subsequently, the eyes were embedded in Technovit 7100 (Kulzer, Wehrheim, Germany) and sectioned (3 μm) as previously described [
39]. Slides were dried, counterstained with 0.5% toluidine blue and mounted under coverslips using Entellan (Merk, Darmstadt, Germany). Eye sections were scanned using a Pannoramic 250 digital slide scanner (3DHISTECH Ltd., Budapest, Hungary) and images were processed with CaseViewer 2.1 (3DHISTECH Ltd., Budapest, Hungary).
4.6. Immunohistochemical Analysis
Eyes were collected at a range of time points from P5 to 3-month-old control and Crb1 mutant rats (n = 2–4/age/group). For immunohistochemical analysis, eyes were enucleated and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Then, the eyes were cryo-protected with 15% and 30% sucrose in PBS, embedded in Tissue-Tek O.C.T Compound (Sakura, Finetek), and used for cryosectioning. Cryosections of 8 µm were made with a Leica CM1900 cryostat (Leica Microsystems).
Sections for immunohistochemistry were blocked for 1 h at RT in 10% normal goat serum, 0.4% Triton X-100 and 1% bovine serum albumin (BSA) in PBS. The primary antibodies were diluted in 0.3% normal goat serum, 0.4% Triton X-100 and 1% BSA in PBS and incubated in a moist chamber overnight at 4 °C. After rinsing in PBS, the sections were incubated for 1h at RT with the fluorescent-labelled secondary antibodies goat anti-mouse, goat anti-rabbit or goat anti-chicken IgGs conjugated to Alexa 488, Alexa 555 (1:1000; Abcam) or Cy3 (1:500), which were diluted in 0.1% goat serum in PBS. Nuclei were counterstained with DAPI and mounted in a Vectashield Hardset mounting medium (H1500 or H1800, Vector Laboratories, Burlingame, USA). Sections were imaged on a Leica TCS SP8 confocal microscope. Confocal images were processed with Leica Application Suite X (v3.7.0.20979).
The following primary antibodies were used: P120-catenin (1:250; BD Biosciences Cat# 610134), CRB1 AK2 (1:200; homemade), CRB2 SK11 (1:200; [
3]), glutamine synthetase (GS) (1:250; BD Biosciences Cat# 610518), PALS1 (1:200; homemade), recoverin (1:500; Millipore Cat# AB5585) and rhodopsin (1:500; Millipore Cat# MAB5356), SOX9 (1:250; Millipore Cat# AB5535).
4.7. Spectral Domain Optical Coherence Tomography (SD-OCT)
P17, 1, 2 and 3 month control and Crb1 mutant rats were anesthetized using 60 mg/kg ketamine and 60 mg/kg xylazine (50 mg/kg ketamine and 5 mg/kg xylazine for P17 rats) intraperitoneally and the pupils were diluted using tropicamide drops (5 mg/mL). Anesthetized rats were placed in front of the SD-OCT imaging device (EnvisuTM R2210 VHR, Leica, USA). Eyes were kept moisturized with Vidisic Carbogel and Systane ultra-eyedrops during the whole procedure. Image acquisitions were performed using the following parameters: rectangular scans of 3.2 mm by 3.2 mm, A-scans/B-scans: 1000, B-scans: 100, Frames/B-scan: 6 (for high resolution B-scans); and A-scans/B-scans: 400, B-scans: 400, Frames/B-scan: 4 (also known as the isotropic scan for an enface projection image). Thickness of retinal layers were manually measured using Bioptigen InVivoVue Reader and Diver software in the individual layers at 0.3, 0.6, 0.9 and 1.3 mm both sides from the center of the optic nerve head in the nasal–temporal direction. In addition, the length of disrupted and healthy retinal lamination was measured using Fiji ImageJ software, where the frame with the optic nerve head and 800 µm before and after the optic nerve head in nasal-temporal directions were used for quantification. Values of the three different frames in the left and right eye were averaged and plotted in the figure together; so, one value per animal.
4.8. Immuno-Electron Microscopy
Immuno-electron microscopy was performed as previously described [
40]. In brief, 40 µm sections were incubated with a primary antibody for 48h, it was then incubated with a secondary peroxidase anti-peroxidase for 4 h. After that, sections were developed in a 2,2-diaminobenzidine solution for 4–8 min, and then the gold-substitute-silver-peroxidase method was applied. Sections were then prepared for electron microscopy and overlapping images were collected using a One View Camera (Gatan) as previously described [
41].
4.9. Delivery of the AAV
For tropism experiments, five days old rats were anesthetized using hypothermia and eight days old rats were anesthetized using an intraperitoneally injected with 35 mg/kg ketamine and 35 mg/kg xylazine. Eyelids were opened and eyes were popped out using surgical tools, the pupils were dilated with 1% tropicamide drops (5 mg/mL) and kept moist with Hypromellose drops. Under visualization with an operating microscope intravitreal and subretinal injections were performed in control and Crb1 mutant rats. We used AAV2/5, AAV2/9 or AAV2/ShH10Y445F, with the full-length CMV promotor, GFP and bovine growth hormone polyadenylation sequence for our tropism study. For each serotype and route of delivery, a dose of ~1 × 1013 gc/mL was injected in a volume of 1 µL using a 33-gauge blunt-tipped Hamilton syringe (Hamilton Company, Reno, NV, USA). Eyes were closed and protected with “Hansaplast liquid protection” and treated with ointment containing chloramphenicol to prevent infections.
For the gene therapy experiments, similar procedures were followed as described above. Here, one eye was treated with an AAV vector and the other eye was mock-injected (PBS); this was randomized in the left and right eye. We used intravitreal injection of ShH10Y capsids to package the AAV2 inverted terminal repeats, the full length or minimal CMV promoter (CMV, or CMVmin), h
CRB1 or h
CRB2 cDNAs and synthetic poly-adenylation sequence for our gene therapy experiments. The AAVs were spiked with 1/10 dose ShH10Y, with the full-length CMV promotor,
GFP cDNA and bovine growth hormone polyadenylation sequence. We carefully examined the quality of our AAV-h
CRB vector preparations by qPCR and Western blots; we recently showed before that these batches of AAV vectors worked efficiently in h
CRB gene therapy studies [
13].
4.10. Tropism Quantification
Immunohistochemical slides for tropism studies were imaged on a Leica TCS SP8 confocal microscope, and sections were scanned for AAV-GFP-positive areas to re-confirm the subretinal or intravitreal injection and define the tropism. For each condition, 4–8 confocal images spanning the area of transduction of each eye was acquired (n ≥ 2 independently injected eyes for each condition). Each individual confocal image was acquired at 40X magnification. The number of eGFP-positive cells within each layer were manually counted. The total number of eGFP-positive cells per eye were divided by transduction diameter to determine the number of positive cells per 100 μm. Then eyes were averaged and coded as - = 0 GFP+ cells; +/−; 1 GFP+ cells, +; 2–5 GFP+ cells, ++; 6–10 GFP+ cells, +++; 11–15 GFP+ cells, ++++; and >16 GFP+ cells.
4.11. Statistical Analysis
We performed statistical analysis for group comparisons: comparing the untreated Crb1 mutant and age-matched control rats by ERG using a two-way ANOVA; comparing all AAV-treated eyes with the other untreated or PBS-injected eyes using a two-way ANOVA (at 2 and 3 months of age); comparing the AAV-treated eye with the other untreated or PBS-injected eyes using a paired t-test; and, finally, statistical analysis on SD-OCT quantifications using a two-way ANOVA. These statistical analyses were performed using GraphPad Prism version 8 (GraphPad Software). All values are expressed as the mean ± SEM if not otherwise indicated. Statistically significant values: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}