
The Interaction of Diet and Mitochondrial Dysfunction in Aging and Cognition

Abstract
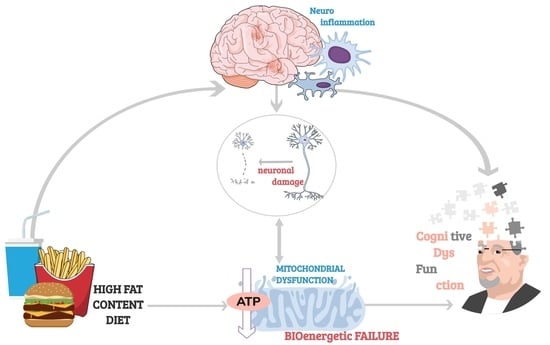
:
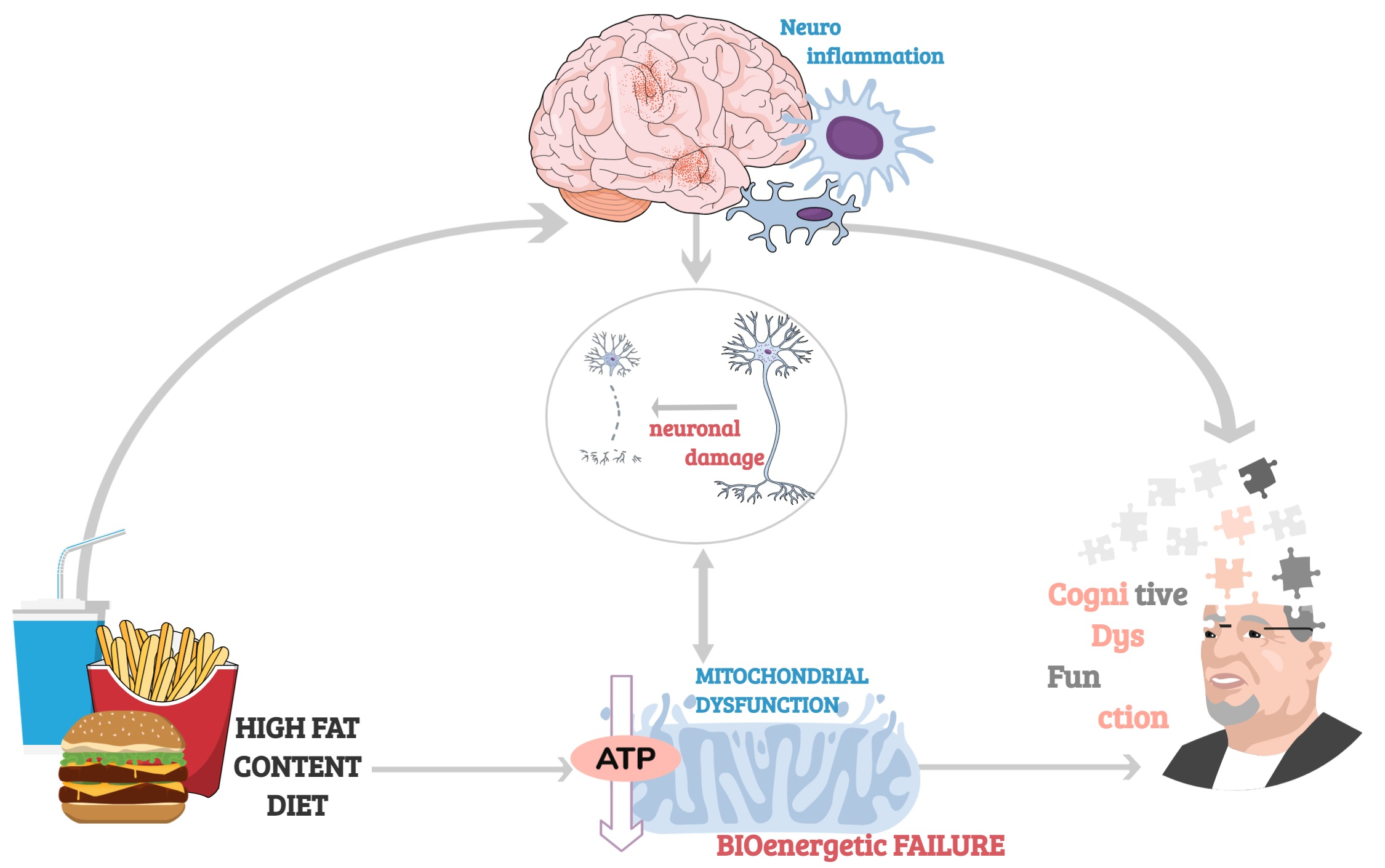{kind=link}
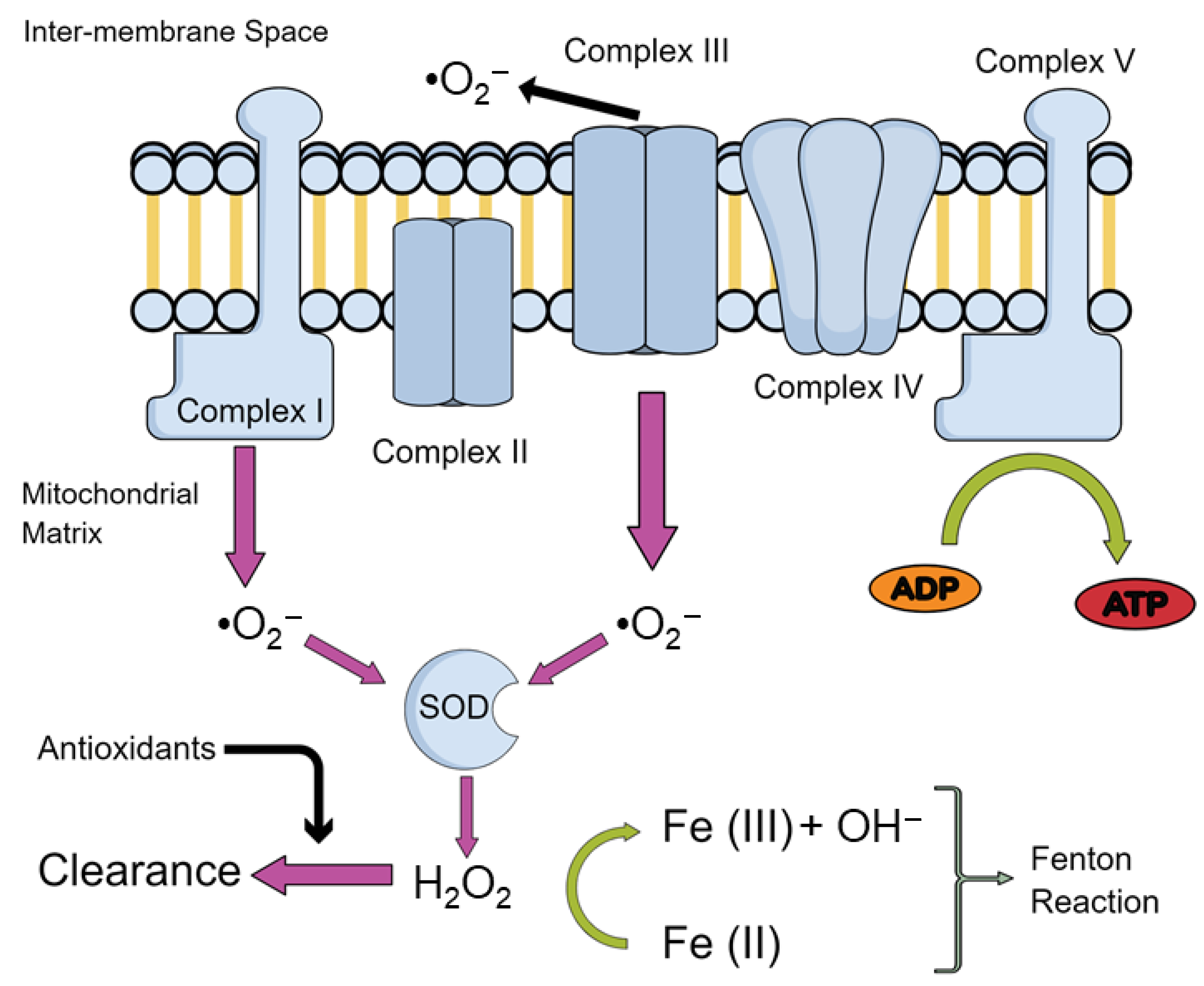{kind=link}
{kind=link}
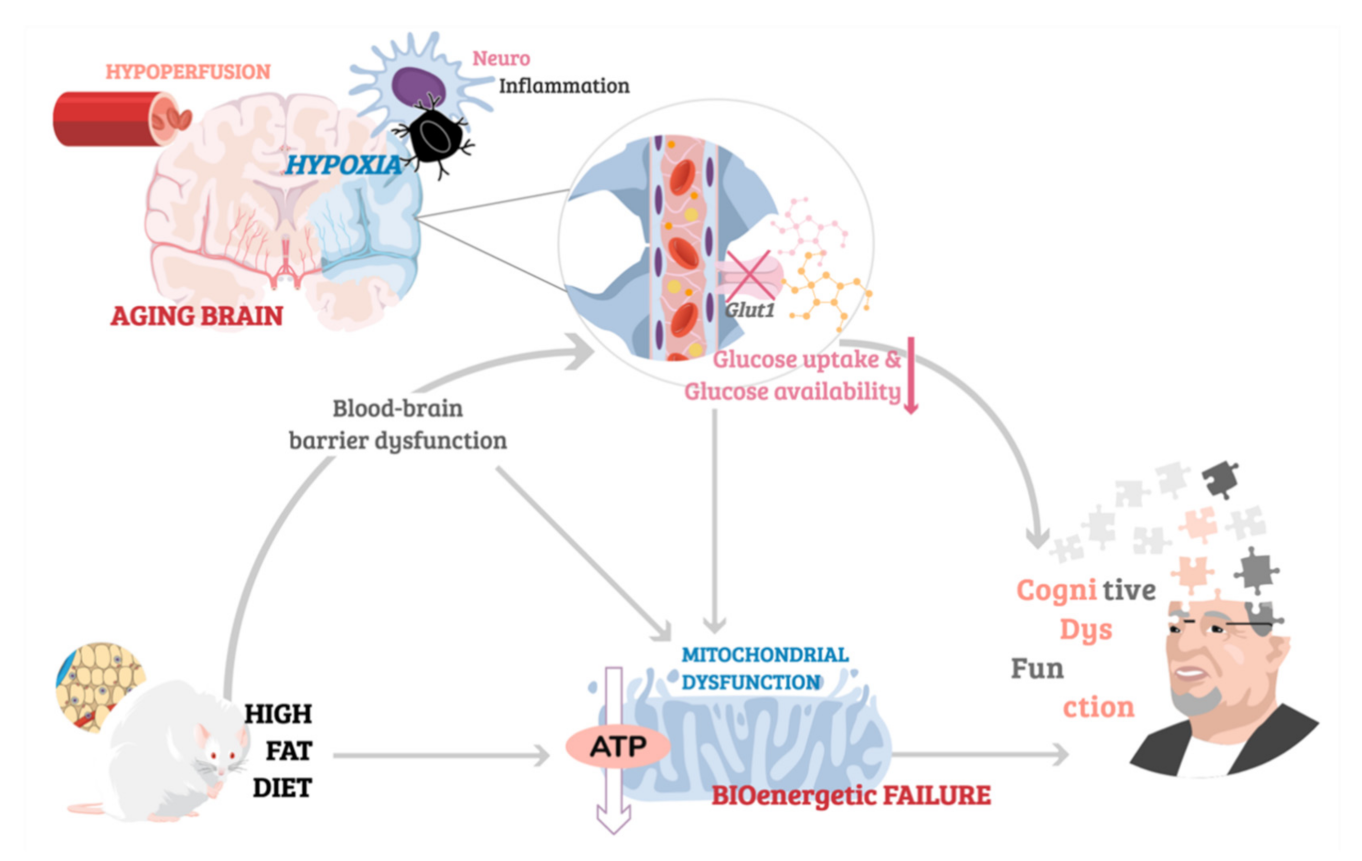{kind=link}
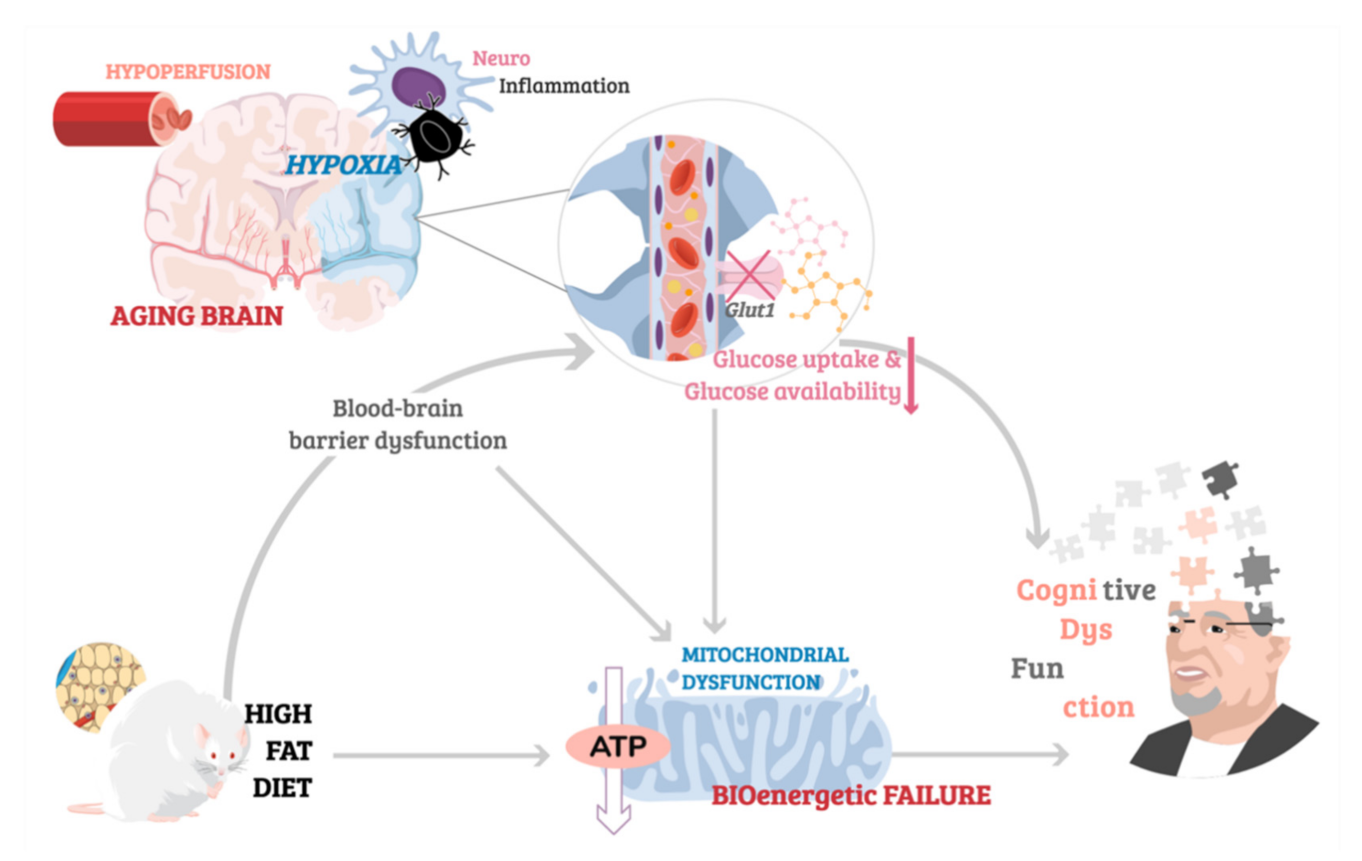
1. Introduction
2. ROS and Its Effects on the Mitochondria
2.1. Oxidative Damage to Lipids and Protein in Aging
2.2. Lipofuscinosis as a Mitochondrial Dysfunction Marker in Aging
2.3. Oxidative Damage to DNA in Aging
3. Mitochondrial Dysfunction and “Inflamm-Aging”
Inflamm-Aging: Inflammatory-Mitochondrial Dysfunction in Aging
4. Mitochondria Dysfunction and Immunity in Aging
5. Mitochondria Dysfunction and Cognitive Impairment in Aging
5.1. Glucose Metabolism and Cognition in Aging
5.2. Breakdown of Mitochondrial Function in Age-Related Cognitive Decline.
5.3. Diet Contribution to Synaptic Dysfunction in Aging
6. The Role of Diet in Aging
Diet Interventions Targetting Aging-Mitochondrial Dysfunction
7. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar] [CrossRef]
- Zhu, X.H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. Neuroimage 2012, 60, 2107–2117. [Google Scholar] [CrossRef] [Green Version]
- Raichle, M.E.; Gusnard, D.A. Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. USA 2002, 99, 10237–10239. [Google Scholar] [CrossRef] [Green Version]
- Palay, S.L. Synapses in the central nervous system. J. Biophys. Biochem. Cytol. 1956, 2, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.L.; Blain, A.P.; Turnbull, D.M.; Gorman, G.S. Systematic review of cognitive deficits in adult mitochondrial disease. Eur. J. Neurol. 2020, 27, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Jezek, P.; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Mackey, M.M.; Diaz, A.A.; Cox, D.P. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: Implications for diseases associated with iron accumulation. Redox Rep. Commun. Free Radic. Res. 2009, 14, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Klann, E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 2007, 9, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Grimm, A.; Friedland, K.; Eckert, A. Mitochondrial dysfunction: The missing link between aging and sporadic Alzheimer’s disease. Biogerontology 2016, 17, 281–296. [Google Scholar] [CrossRef]
- Spencer, S.J.; D’Angelo, H.; Soch, A.; Watkins, L.R.; Maier, S.F.; Barrientos, R.M. High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiol. Aging 2017, 58, 88–101. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef]
- Noronha, S.S.R.; Lima, P.M.; Campos, G.S.V.; Chirico, M.T.T.; Abreu, A.R.; Figueiredo, A.B.; Silva, F.C.S.; Chianca, D.A., Jr.; Lowry, C.A.; De Menezes, R.C.A. Association of high-fat diet with neuroinflammation, anxiety-like defensive behavioral responses, and altered thermoregulatory responses in male rats. Brain Behav. Immun. 2019, 80, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef]
- Vauzour, D.; Camprubi-Robles, M.; Miquel-Kergoat, S.; Andres-Lacueva, C.; Bánáti, D.; Barberger-Gateau, P.; Bowman, G.L.; Caberlotto, L.; Clarke, R.; Hogervorst, E.; et al. Nutrition for the ageing brain: Towards evidence for an optimal diet. Ageing Res. Rev. 2017, 35, 222–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardener, H.; Caunca, M.R. Mediterranean Diet in Preventing Neurodegenerative Diseases. Curr. Nutr. Rep. 2018, 7, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Zamroziewicz, M.K.; Barbey, A.K. The Mediterranean Diet and Healthy Brain Aging: Innovations From Nutritional Cognitive Neuroscience. In Role of the Mediterranean Diet in the Brain and Neurodegenerative Diseases; Farooqui, T., Farooqui, A.A., Eds.; Academic Press: Cambridge, MA, USA, 2018; Chapter 2; pp. 17–33. [Google Scholar] [CrossRef]
- Willett, W.C.; Sacks, F.; Trichopoulou, A.; Drescher, G.; Ferro-Luzzi, A.; Helsing, E.; Trichopoulos, D. Mediterranean diet pyramid: A cultural model for healthy eating. Am. J. Clin. Nutr. 1995, 61, 1402S–1406S. [Google Scholar] [CrossRef] [PubMed]
- Gardener, S.L.; Rainey-Smith, S.R.; Barnes, M.B.; Sohrabi, H.R.; Weinborn, M.; Lim, Y.Y.; Harrington, K.; Taddei, K.; Gu, Y.; Rembach, A.; et al. Dietary patterns and cognitive decline in an Australian study of ageing. Mol. Psychiatry 2015, 20, 860–866. [Google Scholar] [CrossRef]
- Kesse-Guyot, E.; Andreeva, V.A.; Jeandel, C.; Ferry, M.; Hercberg, S.; Galan, P. A healthy dietary pattern at midlife is associated with subsequent cognitive performance. J. Nutr. 2012, 142, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Tangney, C.C.; Kwasny, M.J.; Li, H.; Wilson, R.S.; Evans, D.A.; Morris, M.C. Adherence to a Mediterranean-type dietary pattern and cognitive decline in a community population. Am. J. Clin. Nutr. 2011, 93, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Cherbuin, N.; Anstey, K.J. The Mediterranean diet is not related to cognitive change in a large prospective investigation: The PATH Through Life study. Am. J. Geriatr. Psychiatry 2012, 20, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Haring, B.; Wu, C.; Mossavar-Rahmani, Y.; Snetselaar, L.; Brunner, R.; Wallace, R.B.; Neuhouser, M.L.; Wassertheil-Smoller, S. No Association between Dietary Patterns and Risk for Cognitive Decline in Older Women with 9-Year Follow-Up: Data from the Women’s Health Initiative Memory Study. J. Acad. Nutr. Diet. 2016, 116, 921–930.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samieri, C.; Grodstein, F.; Rosner, B.A.; Kang, J.H.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Willett, W.C.; Okereke, O.I. Mediterranean diet and cognitive function in older age. Epidemiology 2013, 24, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Venkateshappa, C.; Harish, G.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: Implications for neurodegeneration in Alzheimer’s disease. Neurochem. Res. 2012, 37, 1601–1614. [Google Scholar] [CrossRef]
- Mandal, P.K.; Tripathi, M.; Sugunan, S. Brain oxidative stress: Detection and mapping of anti-oxidant marker ‘Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 2012, 417, 43–48. [Google Scholar] [CrossRef]
- Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice. Brain Res. 2007, 1127, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Toman, J.; Fiskum, G. Influence of aging on membrane permeability transition in brain mitochondria. J. Bioenerg Biomembr. 2011, 43, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Fu, M.; Shi, W.; Li, Z.; Liu, H. Activation of mPTP-dependent mitochondrial apoptosis pathway by a novel pan HDAC inhibitor resminostat in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 477, 527–533. [Google Scholar] [CrossRef]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef]
- Ward, R.; Li, W.; Abdul, Y.; Jackson, L.; Dong, G.; Jamil, S.; Filosa, J.; Fagan, S.C.; Ergul, A. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol. Res. 2019, 142, 237–250. [Google Scholar] [CrossRef]
- Hou, B.; Zhang, Y.; Liang, P.; He, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020, 11, 377. [Google Scholar] [CrossRef]
- Rottenberg, H.; Wu, S. Mitochondrial dysfunction in lymphocytes from old mice: Enhanced activation of the permeability transition. Biochem. Biophys. Res. Commun. 1997, 240, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Borst, S.E.; Conover, C.F. High-fat diet induces increased tissue expression of TNF-alpha. Life Sci. 2005, 77, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Sana, R.; Calder, V.; Calonge, M.; Lee, W.; Wheeler, L.A.; Stern, M.E. Mitochondrial permeability transition pore in inflammatory apoptosis of human conjunctival epithelial cells and T cells: Effect of cyclosporin A. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4717–4733. [Google Scholar] [CrossRef] [PubMed]
- Capaldi, R.A. Arrangement of proteins in the mitochondrial inner membrane. Biochim. Biophys. Acta 1982, 694, 291–306. [Google Scholar] [CrossRef]
- Robinson, N.C. Functional binding of cardiolipin to cytochrome c oxidase. J. Bioenergy Biomembr. 1993, 25, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hoch, F.L. Cardiolipins and biomembrane function. Biochim. Biophys. Acta 1992, 1113, 71–133. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Hostetler, K.Y. Mammalian cardiolipin biosynthesis. Methods Enzym. 1992, 209, 330–337. [Google Scholar] [CrossRef]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci 2008, 65, 2493–2506. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: Role of cardiolipin. FEBS Lett. 1997, 406, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Petrosillo, G.; Ruggiero, F.M. Cardiolipin-dependent decrease of cytochrome c oxidase activity in heart mitochondria from hypothyroid rats. Biochim. Biophys. Acta 1997, 1319, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Singer, T.P.; Ramsay, R.R.; Ackrell, B.A. Deficiencies of NADH and succinate dehydrogenases in degenerative diseases and myopathies. Biochim. Biophys. Acta 1995, 1271, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Forsmark-Andree, P.; Lee, C.P.; Dallner, G.; Ernster, L. Lipid peroxidation and changes in the ubiquinone content and the respiratory chain enzymes of submitochondrial particles. Free Radic. Biol. Med. 1997, 22, 391–400. [Google Scholar] [CrossRef]
- Lippe, G.; Comelli, M.; Mazzilis, D.; Sala, F.D.; Mavelli, I. The inactivation of mitochondrial F1 ATPase by H2O2 is mediated by iron ions not tightly bound in the protein. Biochem. Biophys. Res. Commun. 1991, 181, 764–770. [Google Scholar] [CrossRef]
- Lippe, G.; Londero, D.; Sala, F.D.; Mavelli, I. H2O2-induced damage to beef heart mitochondria F0F1 ATP synthase complex: Differential sensitivity of the F1 and F0 moieties. Biochem. Mol. Biol. Int. 1993, 30, 1061–1070. [Google Scholar] [PubMed]
- Desai, I.D.; Tappel, A.L. Damage to proteins by peroxidized lipids. J. Lipid Res. 1963, 4, 204–207. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, P.; Liu, R.; Li, Y.; Liu, C.; Liao, X. A DEHP plasticizer alters synaptic proteins via peroxidation. Toxicol. Res. 2017, 6, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic. Biol. Med. 2019, 130, 140–150. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Peroxidative damage to cardiac mitochondria: Cytochrome oxidase and cardiolipin alterations. FEBS Lett. 1998, 424, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Keyer, K.; Imlay, J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Strehler, B.L.; Mark, D.D.; Mildvan, A.S. GEE MV: Rate and magnitude of age pigment accumulation in the human myocardium. J. Gerontol. 1959, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, M.R.; Greenwood, J.G.; Fielder, D.R. Lipofuscin as a record of “rate of living” in an aquatic poikilotherm. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, B327–B336. [Google Scholar] [CrossRef]
- Hosokawa, H.; Ishii, N.; Ishida, H.; Ichimori, K.; Nakazawa, H.; Suzuki, K. Rapid accumulation of fluorescent material with aging in an oxygen-sensitive mutant mev-1 of Caenorhabditis elegans. Mech. Ageing Dev. 1994, 74, 161–170. [Google Scholar] [CrossRef]
- Nakano, M.; Mizuno, T.; Gotoh, S. Accumulation of cardiac lipofuscin in crab-eating monkeys (Macaca fasicularis): The same rate of lipofuscin accumulation in several species of primates. Mech. Ageing Dev. 1993, 66, 243–248. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Lipofuscin: Mechanisms of formation and increase with age. APMIS 1998, 106, 265–276. [Google Scholar] [CrossRef]
- Elleder, M.; Sokolova, J.; Hrebicek, M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol. 1997, 93, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.Y.; Su, Y.B.; Zhang, F.R.; Li, J.F. Effects of vitamin E on the blastogenic response of splenocytes and lipofuscin contents in the hearts and brains of aged mice. J. Environ. Pathol. Toxicol. Oncol. 1996, 15, 51–53. [Google Scholar] [PubMed]
- Ohki, K.; Takamura, T.; Nozawa, Y. Effect of alpha-tocopherol on lipid peroxidation and acyl chain mobility of liver microsomes from vitamin E-difficient rat. J. Nutr. Sci. Vitam. 1984, 30, 221–234. [Google Scholar] [CrossRef]
- Palmer, D.N. The relevance of the storage of subunit c of ATP synthase in different forms and models of Batten disease (NCLs). Biochim. Biophys. Acta 2015, 1852, 2287–2291. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.L.; Gao, C.L.; Tompkins, J.A.; Bronson, R.T.; Chin, D.T. Mitochondrial ATP synthase subunit c stored in hereditary ceroid-lipofuscinosis contains trimethyl-lysine. Biochem. J. 1995, 310 Pt 3, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Hwang, W.; Jeong, D.E.; Ryu, Y.; Ha, C.M.; Lee, S.V.; Liu, L.; He, Z.M. Genetic inhibition of an ATP synthase subunit extends lifespan in C. elegans. Sci. Rep. 2018, 8, 14836. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Dizdaroglu, M. Chemical determination of free radical-induced damage to DNA. Free Radic. Biol. Med. 1991, 10, 225–242. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Ames, B.N. Assays for 8-hydroxy-2′-deoxyguanosine: A biomarker of in vivo oxidative DNA damage. Free Radic. Biol. Med. 1991, 10, 211–216. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, L.; Bialkowski, K.; Risom, L.; Lohr, M.; Loft, S.; Moller, P. Aging and defense against generation of 8-oxo-7,8-dihydro-2′-deoxyguanosine in DNA. Free Radic. Biol. Med. 2009, 47, 608–615. [Google Scholar] [CrossRef]
- Souza-Pinto, N.C.; Croteau, D.L.; Hudson, E.K.; Hansford, R.G.; Bohr, V.A. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999, 27, 1935–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gredilla, R.; Garm, C.; Holm, R.; Bohr, V.A.; Stevnsner, T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. Aging 2010, 31, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Vyjayanti, V.N.; Rao, K.S. DNA double strand break repair in brain: Reduced NHEJ activity in aging rat neurons. Neurosci. Lett. 2006, 393, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Herzig, M.; Rotrekl, V.; Walter, C.A. Base excision repair, aging and health span. Mech. Ageing Dev. 2008, 129, 366–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Juhaszova, M.; Sollott, S.J. Mitochondrial health, the epigenome and healthspan. Clin. Sci. 2016, 130, 1285–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M. Neurodegeneration in accelerated aging. Dan. Med. J. 2016, 63, B5308. [Google Scholar] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Richter, T.; von Zglinicki, T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 2007, 42, 1039–1042. [Google Scholar] [CrossRef]
- Serra, V.; von Zglinicki, T.; Lorenz, M.; Saretzki, G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J. Biol. Chem. 2003, 278, 6824–6830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalai, V.A.; Singer, M.J.; Thorp, H.H. Site-specific probing of oxidative reactivity and telomerase function using 7,8-dihydro-8-oxoguanine in telomeric DNA. J. Am. Chem. Soc. 2002, 124, 1625–1631. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyolfson, E.; Malik, H.; Mychasiuk, R. Sexually Dimorphic Behavioral and Genetic Outcomes Associated With Administration of TA65 (A Telomerase Activator) Following Repetitive Traumatic Brain Injury: A Pilot Study. Front. Neurol. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardes de Jesus, B.; Schneeberger, K.; Vera, E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavretsky, H.; Epel, E.S.; Siddarth, P.; Nazarian, N.; Cyr, N.S.; Khalsa, D.S.; Lin, J.; Blackburn, E.; Irwin, M.R. A pilot study of yogic meditation for family dementia caregivers with depressive symptoms: Effects on mental health, cognition, and telomerase activity. Int. J. Geriatr. Psychiatry 2013, 28, 57–65. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadinanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M., 3rd; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Tormos, A.M.; Lerner, C.A.; Gerloff, J.; Yao, H.; Rahman, I. Shelterin Telomere Protection Protein 1 Reduction Causes Telomere Attrition and Cellular Senescence via Sirtuin 1 Deacetylase in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2017, 56, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Hohensinner, P.J.; Kaun, C.; Buchberger, E.; Ebenbauer, B.; Demyanets, S.; Huk, I.; Eppel, W.; Maurer, G.; Huber, K.; Wojta, J. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim. Biophys. Acta 2016, 1863, 360–367. [Google Scholar] [CrossRef]
- Achanta, G.; Huang, P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004, 64, 6233–6239. [Google Scholar] [CrossRef] [Green Version]
- Artandi, S.E.; Attardi, L.D. Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochem. Biophys. Res. Commun. 2005, 331, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Horikawa, I.; Mondal, A.M.; Jenkins, L.M.; Appella, E.; Vojtesek, B.; Bourdon, J.C.; Lane, D.P.; Harris, C.C. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat. Cell Biol. 2010, 12, 1205–1212. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, I.; Fujita, K.; Harris, C.C. p53 governs telomere regulation feedback too, via TRF2. Aging 2011, 3, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Oettinghaus, B.; Schulz, J.M.; Restelli, L.M.; Licci, M.; Savoia, C.; Schmidt, A.; Schmitt, K.; Grimm, A.; Morè, L.; Hench, J.; et al. Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 2016, 23, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Xiong, H.; Su, Z.; Pang, J.; Lai, L.; Zhang, H.; Jian, B.; Zhang, W.; Zheng, Y. Inhibition of DRP-1-Dependent Mitophagy Promotes Cochlea Hair Cell Senescence and Exacerbates Age-Related Hearing Loss. Front. Cell. Neurosci. 2019, 13, 550. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappaB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerondakis, S.; Grumont, R.; Rourke, I.; Grossmann, M. The regulation and roles of Rel/NF-kappa B transcription factors during lymphocyte activation. Curr. Opin. Immunol. 1998, 10, 353–359. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Cho, J.A.; Kim, T.J.; Moon, H.J.; Kim, Y.J.; Yoon, H.K.; Seong, S.Y. Cardiolipin activates antigen-presenting cells via TLR2-PI3K-PKN1-AKT/p38-NF-kB signaling to prime antigen-specific naïve T cells in mice. Eur. J. Immunol. 2018, 48, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Gemechu, J.M.; Bentivoglio, M. T Cell Recruitment in the Brain during Normal Aging. Front. Cell. Neurosci. 2012, 6, 38. [Google Scholar] [CrossRef] [Green Version]
- Batterman, K.V.; Cabrera, P.E.; Moore, T.L.; Rosene, D.L. T Cells Actively Infiltrate the White Matter of the Aging Monkey Brain in Relation to Increased Microglial Reactivity and Cognitive Decline. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, P.J. Mitochondrial Dysfunction and the Aging Immune System. Biology 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, E.A.; Lum, L.G.; L’Hommedieu, G.; Goodwin, J.S. Specific humoral immunity in the elderly: In vivo and in vitro response to vaccination. J. Gerontol. 1993, 48, B231–B236. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Chapman, A.J.; Goree, A.; Jonsson, S.; Briles, D.; Baughn, R.E. Natural and vaccine-related immunity to Streptococcus pneumoniae. J. Infect. Dis. 1986, 154, 245–256. [Google Scholar] [CrossRef]
- van der Windt, G.J.; O’Sullivan, D.; Everts, B.; Huang, S.C.; Buck, M.D.; Curtis, J.D.; Chang, C.H.; Smith, A.M.; Ai, T.; Faubert, B.; et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Tarasenko, T.N.; Pacheco, S.E.; Koenig, M.K.; Gomez-Rodriguez, J.; Kapnick, S.M.; Diaz, F.; Zerfas, P.M.; Barca, E.; Sudderth, J.; DeBerardinis, R.J.; et al. Cytochrome c Oxidase Activity Is a Metabolic Checkpoint that Regulates Cell Fate Decisions During T Cell Activation and Differentiation. Cell Metab. 2017, 25, 1254–1268.e57. [Google Scholar] [CrossRef] [Green Version]
- Callender, L.A.; Carroll, E.C.; Bober, E.A.; Akbar, A.N.; Solito, E.; Henson, S.M. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 2020, 19, e13067. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.G.; Ding, J.Q.; Chen, S.D. Microglia in the aging brain: Relevance to neurodegeneration. Mol. Neurodegener. 2010, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflamm. 2014, 11, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Spencer, S.J.; Basri, B.; Sominsky, L.; Soch, A.; Ayala, M.T.; Reineck, P.; Gibson, B.C.; Barrientos, R.M. High-fat diet worsens the impact of aging on microglial function and morphology in a region-specific manner. Neurobiol. Aging 2019, 74, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Nadjar, A. Role of metabolic programming in the modulation of microglia phagocytosis by lipids. Prostaglandins Leukot Essent Fatty Acids 2018, 135, 63–73. [Google Scholar] [CrossRef]
- Fjell, A.M.; Walhovd, K.B.; Fennema-Notestine, C.; McEvoy, L.K.; Hagler, D.J.; Holland, D.; Brewer, J.B.; Dale, A.M. One-year brain atrophy evident in healthy aging. J. Neurosci. 2009, 29, 15223–15231. [Google Scholar] [CrossRef]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Dore, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef]
- Tarumi, T.; Zhang, R. Cerebral blood flow in normal aging adults: Cardiovascular determinants, clinical implications, and aerobic fitness. J. Neurochem. 2018, 144, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J.; Rosas, H.D.; Salat, D.H. Age-associated reductions in cerebral blood flow are independent from regional atrophy. Neuroimage 2011, 55, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Strandgaard, S. Cerebral blood flow in the elderly: Impact of hypertension and antihypertensive treatment. Cardiovasc. Drugs 1991, 4 (Suppl. 6), 1217–1221. [Google Scholar] [CrossRef]
- Shaw, T.G.; Mortel, K.F.; Meyer, J.S.; Rogers, R.L.; Hardenberg, J.; Cutaia, M.M. Cerebral blood flow changes in benign aging and cerebrovascular disease. Neurology 1984, 34, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Galiano, A.; Mengual, E.; García de Eulate, R.; Galdeano, I.; Vidorreta, M.; Recio, M.; Riverol, M.; Zubieta, J.L.; Fernández-Seara, M.A. Coupling of cerebral blood flow and functional connectivity is decreased in healthy aging. Brain Imaging Behav. 2020, 14, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Oz, G.; Seaquist, E.R.; Kumar, A.; Criego, A.B.; Benedict, L.E.; Rao, J.P.; Henry, P.G.; Van De Moortele, P.F.; Gruetter, R. Human brain glycogen content and metabolism: Implications on its role in brain energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E946–E951. [Google Scholar] [CrossRef]
- Verclytte, S.; Lopes, R.; Lenfant, P.; Rollin, A.; Semah, F.; Leclerc, X.; Pasquier, F.; Delmaire, C. Cerebral Hypoperfusion and Hypometabolism Detected by Arterial Spin Labeling MRI and FDG-PET in Early-Onset Alzheimer’s Disease. J. Neuroimaging 2016, 26, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Sabri, O.; Ringelstein, E.B.; Hellwig, D.; Schneider, R.; Schreckenberger, M.; Kaiser, H.J.; Mull, M.; Buell, U. Neuropsychological impairment correlates with hypoperfusion and hypometabolism but not with severity of white matter lesions on MRI in patients with cerebral microangiopathy. Stroke 1999, 30, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.S.; Rauch, G.; Rauch, R.A.; Haque, A. Risk factors for cerebral hypoperfusion, mild cognitive impairment, and dementia. Neurobiol. Aging 2000, 21, 161–169. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 943–972. [Google Scholar] [CrossRef]
- Magaki, S.; Yong, W.H.; Khanlou, N.; Tung, S.; Vinters, H.V. Comorbidity in dementia: Update of an ongoing autopsy study. J. Am. Geriatr. Soc. 2014, 62, 1722–1728. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.N.; Tan, C.C.; Shen, X.N.; Xu, W.; Hou, X.H.; Dong, Q.; Tan, L.; Yu, J.T. Blood Pressure and Risks of Cognitive Impairment and Dementia: A Systematic Review and Meta-Analysis of 209 Prospective Studies. Hypertension 2020, 76, 217–225. [Google Scholar] [CrossRef]
- Sekaran, H.; Gan, C.Y.; Latiff, A.A.; Harvey, T.M.; Mohd Nazri, L.; Hanapi, N.A.; Azizi, J.; Yusof, S.R. Changes in blood-brain barrier permeability and ultrastructure, and protein expression in a rat model of cerebral hypoperfusion. Brain Res. Bull. 2019, 152, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.M.; Jansen, J.F.A.; Zhang, C.E.; Hoff, E.I.; Staals, J.; van Oostenbrugge, R.J.; Backes, W.H. Blood-brain barrier impairment and hypoperfusion are linked in cerebral small vessel disease. Neurology 2019, 92, e1669–e1677. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, W.; Jia, L.; Qin, W.; Hou, T.; Wu, Q.; Li, H.; Tian, Y.; Jia, J. The Effect of Chronic Cerebral Hypoperfusion on Blood-Brain Barrier Permeability in a Transgenic Alzheimer’s Disease Mouse Model (PS1V97L). J. Alzheimer’s Dis. 2020, 74, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Simon, M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010, 345, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merelli, A.; Rodríguez, J.C.G.; Folch, J.; Regueiro, M.R.; Camins, A.; Lazarowski, A. Understanding the Role of Hypoxia Inducible Factor During Neurodegeneration for New Therapeutics Opportunities. Curr. Neuropharmacol. 2018, 16, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.; Park, J.W.; Hwang, J.S.; Kim, S.M.; Lyoo, I.K.; Lee, C.J.; Han, I.O. Hypoxia-Induced Neuroinflammation and Learning-Memory Impairments in Adult Zebrafish Are Suppressed by Glucosamine. Mol. Neurobiol. 2018, 55, 8738–8753. [Google Scholar] [CrossRef]
- Sapin, E.; Peyron, C.; Roche, F.; Gay, N.; Carcenac, C.; Savasta, M.; Levy, P.; Dematteis, M. Chronic Intermittent Hypoxia Induces Chronic Low-Grade Neuroinflammation in the Dorsal Hippocampus of Mice. Sleep 2015, 38, 1537–1546. [Google Scholar] [CrossRef]
- Aviles-Reyes, R.X.; Angelo, M.F.; Villarreal, A.; Rios, H.; Lazarowski, A.; Ramos, A.J. Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: Implications for sleep apnea. J. Neurochem. 2010, 112, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Lee, J.C.; Yoo, K.Y.; Choi, J.H.; Hwang, I.K.; Cho, J.H.; Kwon, Y.G.; et al. Effects of high-fat diet on neuronal damage, gliosis, inflammatory process and oxidative stress in the hippocampus induced by transient cerebral ischemia. Neurochem. Res. 2014, 39, 2465–2478. [Google Scholar] [CrossRef] [PubMed]
- Stradecki-Cohan, H.M.; Cohan, C.H.; Raval, A.P.; Dave, K.R.; Reginensi, D.; Gittens, R.A.; Youbi, M.; Perez-Pinzon, M.A. Cognitive Deficits after Cerebral Ischemia and Underlying Dysfunctional Plasticity: Potential Targets for Recovery of Cognition. J. Alzheimer’s Dis. 2017, 60, S87–S105. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.; Beaud, V.; Eskandari, A.; Maeder, P.; Demonet, J.F.; Eskioglou, E. Ischemic Amnesia: Causes and Outcome. Stroke 2017, 48, 2270–2273. [Google Scholar] [CrossRef]
- Beatty, W.W.; Salmon, D.P.; Bernstein, N.; Martone, M.; Lyon, L.; Butters, N. Procedural learning in a patient with amnesia due to hypoxia. Brain Cogn. 1987, 6, 386–402. [Google Scholar] [CrossRef]
- Block, F. Global ischemia and behavioural deficits. Prog. Neurobiol. 1999, 58, 279–295. [Google Scholar] [CrossRef]
- Braga, M.M.; Rico, E.P.; Córdova, S.D.; Pinto, C.B.; Blaser, R.E.; Dias, R.D.; Rosemberg, D.B.; Oliveira, D.L.; Souza, D.O. Evaluation of spontaneous recovery of behavioral and brain injury profiles in zebrafish after hypoxia. Behav. Brain Res. 2013, 253, 145–151. [Google Scholar] [CrossRef]
- Shukitt-Hale, B.; Stillman, M.J.; Lieberman, H.R. Tyrosine administration prevents hypoxia-induced decrements in learning and memory. Physiol. Behav. 1996, 59, 867–871. [Google Scholar] [CrossRef]
- Jalal, F.Y.; Yang, Y.; Thompson, J.F.; Roitbak, T.; Rosenberg, G.A. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. J. Cereb. Blood Flow Metab. 2015, 35, 1145–1153. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Oka, M.; Suzuki, E.; Asada, A.; Saito, T.; Iijima, K.M.; Ando, K. Increasing neuronal glucose uptake attenuates brain aging and promotes life span under dietary restriction in Drosophila. iScience 2021, 24, 101979. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yin, F.; Yao, J.; Brinton, R.D.; Cadenas, E. Lipoic acid restores age-associated impairment of brain energy metabolism through the modulation of Akt/JNK signaling and PGC1α transcriptional pathway. Aging Cell 2013, 12, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Pardo, J.V.; Lee, J.T.; Sheikh, S.A.; Surerus-Johnson, C.; Shah, H.; Munch, K.R.; Carlis, J.V.; Lewis, S.M.; Kuskowski, M.A.; Dysken, M.W. Where the brain grows old: Decline in anterior cingulate and medial prefrontal function with normal aging. Neuroimage 2007, 35, 1231–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salat, D.H.; Buckner, R.L.; Snyder, A.Z.; Greve, D.N.; Desikan, R.S.; Busa, E.; Morris, J.C.; Dale, A.M.; Fischl, B. Thinning of the cerebral cortex in aging. Cereb. Cortex 2004, 14, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elahi, D.; Muller, D.C. Carbohydrate metabolism in the elderly. Eur. J. Clin. Nutr. 2000, 54 (Suppl. 3), S112–S120. [Google Scholar] [CrossRef] [Green Version]
- Wennberg, A.M.; Spira, A.P.; Pettigrew, C.; Soldan, A.; Zipunnikov, V.; Rebok, G.W.; Roses, A.D.; Lutz, M.W.; Miller, M.M.; Thambisetty, M.; et al. Blood glucose levels and cortical thinning in cognitively normal, middle-aged adults. J. Neurol. Sci. 2016, 365, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e426. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Tsui, W.H.; Pupi, A.; De Santi, S.; Drzezga, A.; Minoshima, S.; de Leon, M.J. (18)F-FDG PET database of longitudinally confirmed healthy elderly individuals improves detection of mild cognitive impairment and Alzheimer’s disease. J. Nucl. Med. 2007, 48, 1129–1134. [Google Scholar] [CrossRef] [Green Version]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.; Morris, J.C.; Raichle, M.E. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360.e3. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahren, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53 (Suppl. 3), S215–S219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucsek, Z.; Toth, P.; Tarantini, S.; Sosnowska, D.; Gautam, T.; Warrington, J.P.; Giles, C.B.; Wren, J.D.; Koller, A.; Ballabh, P.; et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Ogata, S.; Ito, S.; Masuda, T.; Ohtsuki, S. Changes of Blood-Brain Barrier and Brain Parenchymal Protein Expression Levels of Mice under Different Insulin-Resistance Conditions Induced by High-Fat Diet. Pharm. Res. 2019, 36, 141. [Google Scholar] [CrossRef]
- Nerurkar, P.V.; Johns, L.M.; Buesa, L.M.; Kipyakwai, G.; Volper, E.; Sato, R.; Shah, P.; Feher, D.; Williams, P.G.; Nerurkar, V.R. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J Neuroinflamm. 2011, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell 2016, 165, 882–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, D.; Mitra Mazumder, P. High fat diet administration leads to the mitochondrial dysfunction and selectively alters the expression of class 1 GLUT protein in mice. Mol. Biol. Rep. 2019, 46, 1727–1736. [Google Scholar] [CrossRef]
- Kesby, J.P.; Kim, J.J.; Scadeng, M.; Woods, G.; Kado, D.M.; Olefsky, J.M.; Jeste, D.V.; Achim, C.L.; Semenova, S. Spatial Cognition in Adult and Aged Mice Exposed to High-Fat Diet. PLoS ONE 2015, 10, e0140034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanoski, S.E.; Davidson, T.L. Different patterns of memory impairments accompany short- and longer-term maintenance on a high-energy diet. J. Exp. Psychol. Anim. Behav. Process 2010, 36, 313–319. [Google Scholar] [CrossRef]
- Krishna, S.; Lin, Z.; de La Serre, C.B.; Wagner, J.J.; Harn, D.H.; Pepples, L.M.; Djani, D.M.; Weber, M.T.; Srivastava, L.; Filipov, N.M. Time-dependent behavioral, neurochemical, and metabolic dysregulation in female C57BL/6 mice caused by chronic high-fat diet intake. Physiol. Behav. 2016, 157, 196–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.R.; Chen, Y.; McMullen, M.F.; et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, A.; Hasegawa-Ishii, S. Senescence-accelerated Mice (SAMs) as a Model for Brain Aging and Immunosenescence. Aging Dis. 2011, 2, 414–435. [Google Scholar]
- Nomura, Y.; Yamanaka, Y.; Kitamura, Y.; Arima, T.; Ohnuki, T.; Oomura, Y.; Sasaki, K.; Nagashima, K.; Ihara, Y. Senescence-accelerated mouse. Neurochemical studies on aging. Ann. N. Y. Acad. Sci. 1996, 786, 410–418. [Google Scholar] [CrossRef]
- Kondziella, D.; Hammer, J.; Sletvold, O.; Sonnewald, U. The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: Glial-neuronal metabolic interactions. Neurochem. Int. 2003, 43, 629–637. [Google Scholar] [CrossRef]
- Reutzel, M.; Grewal, R.; Dilberger, B.; Silaidos, C.; Joppe, A.; Eckert, G.P. Cerebral Mitochondrial Function and Cognitive Performance during Aging: A Longitudinal Study in NMRI Mice. Oxid. Med. Cell. Longev. 2020, 2020, 4060769. [Google Scholar] [CrossRef] [Green Version]
- Bowling, A.C.; Mutisya, E.M.; Walker, L.C.; Price, D.L.; Cork, L.C.; Beal, M.F. Age-dependent impairment of mitochondrial function in primate brain. J. Neurochem. 1993, 60, 1964–1967. [Google Scholar] [CrossRef]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.M.; Blithikioti, C.; Ku, M.; et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, T.; Asada, S.; Nishitani, S.; Hazeki, O. Age-related changes in manganese superoxide dismutase activity in the cerebral cortex of senescence-accelerated prone and resistant mouse. Neurosci. Lett. 2001, 298, 135–138. [Google Scholar] [CrossRef]
- Shi, C.; Xiao, S.; Liu, J.; Guo, K.; Wu, F.; Yew, D.T.; Xu, J. Ginkgo biloba extract EGb761 protects against aging-associated mitochondrial dysfunction in platelets and hippocampi of SAMP8 mice. Platelets 2010, 21, 373–379. [Google Scholar] [CrossRef]
- Xu, J.; Shi, C.; Li, Q.; Wu, J.; Forster, E.L.; Yew, D.T. Mitochondrial dysfunction in platelets and hippocampi of senescence-accelerated mice. J. Bioenergy Biomembr. 2007, 39, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lei, H.; Hou, J.; Liu, J. Involvement of oxidative stress in SAMP10 mice with age-related neurodegeneration. Neurol. Sci. 2015, 36, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Pikhart, H.; Mishra, G.; Stephen, A.; Kuh, D.; Richards, M. The role of lifestyle behaviors on 20-year cognitive decline. J. Aging Res. 2012, 2012, 304014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Lu, T.; Loerch, P. The aging brain. Annu. Rev. Pathol. 2008, 3, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.R.; Haley-Zitlin, V.; Stevens, C.; Granholm, A.C. Diet-induced effects on neuronal and glial elements in the middle-aged rat hippocampus. Nutr. Neurosci. 2011, 14, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Dickstein, D.L.; Kabaso, D.; Rocher, A.B.; Luebke, J.I.; Wearne, S.L.; Hof, P.R. Changes in the structural complexity of the aged brain. Aging Cell 2007, 6, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Linville, M.C.; Tucker, E.W.; Sonntag, W.E.; Brunso-Bechtold, J.K. Differential effects of aging and insulin-like growth factor-1 on synapses in CA1 of rat hippocampus. Cereb. Cortex 2005, 15, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Baxter, M.G.; Frick, K.M.; Price, D.L.; Breckler, S.J.; Markowska, A.L.; Gorman, L.K. Presynaptic markers of cholinergic function in the rat brain: Relationship with age and cognitive status. Neuroscience 1999, 89, 771–779. [Google Scholar] [CrossRef]
- Gavilan, M.P.; Revilla, E.; Pintado, C.; Castano, A.; Vizuete, M.L.; Moreno-Gonzalez, I.; Baglietto-Vargas, D.; Sanchez-Varo, R.; Vitorica, J.; Gutierrez, A.; et al. Molecular and cellular characterization of the age-related neuroinflammatory processes occurring in normal rat hippocampus: Potential relation with the loss of somatostatin GABAergic neurons. J. Neurochem. 2007, 103, 984–996. [Google Scholar] [CrossRef]
- Hwang, I.K.; Yoo, K.Y.; Jung, B.K.; Cho, J.H.; Kim, D.H.; Kang, T.C.; Kwon, Y.G.; Kim, Y.S.; Won, M.H. Correlations between neuronal loss, decrease of memory, and decrease expression of brain-derived neurotrophic factor in the gerbil hippocampus during normal aging. Exp. Neurol. 2006, 201, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Majdi, M.; Ribeiro-da-Silva, A.; Cuello, A.C. Cognitive impairment and transmitter-specific pre- and postsynaptic changes in the rat cerebral cortex during ageing. Eur. J. Neurosci. 2007, 26, 3583–3596. [Google Scholar] [CrossRef] [PubMed]
- Mora, F.; Segovia, G.; Del Arco, A. Glutamate-dopamine-GABA interactions in the aging basal ganglia. Brain Res. Rev. 2008, 58, 340–353. [Google Scholar] [CrossRef]
- Shi, L.; Adams, M.M.; Linville, M.C.; Newton, I.G.; Forbes, M.E.; Long, A.B.; Riddle, D.R.; Brunso-Bechtold, J.K. Caloric restriction eliminates the aging-related decline in NMDA and AMPA receptor subunits in the rat hippocampus and induces homeostasis. Exp. Neurol. 2007, 206, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.E.; Majewska, A.K. A role for microglia in synaptic plasticity? Commun. Integr. Biol. 2011, 4, 220–222. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Kuzniewska, B.; Rejmak, E.; Malik, A.R.; Jaworski, J.; Kaczmarek, L.; Kalita, K. Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol. Cell. Biol. 2013, 33, 2149–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139 (Suppl. 2), 91–114. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, J.G.; Yocum, G.T.; Lee, S.S.; Granat, A.; Mikami, M.; Connolly, E.S., Jr.; Heyer, E.J. MMP-9 levels in elderly patients with cognitive dysfunction after carotid surgery. J. Clin. Neurosci. 2010, 17, 436–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Bu, X.; Xu, T.; Guo, L.; Wang, X.; Zhang, J.; Cui, Y.; Li, D.; Zhang, J.; Ju, Z.; et al. Serum Matrix Metalloproteinase-9 and Cognitive Impairment After Acute Ischemic Stroke. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Chiba, Y.; Hattori, S.; Yoshimi, A.; Asami, T.; Katsuse, O.; Suda, A.; Hishimoto, A. Influence of plasma matrix metalloproteinase levels on longitudinal changes in Alzheimer’s disease (AD) biomarkers and cognitive function in patients with mild cognitive impairment due to AD registered in the Alzheimer’s Disease Neuroimaging Initiative database. J. Neurol. Sci. 2020, 416, 116989. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Prakash, R.S.; Voss, M.W.; Chaddock, L.; Heo, S.; McLaren, M.; Pence, B.D.; Martin, S.A.; Vieira, V.J.; Woods, J.A.; et al. Brain-derived neurotrophic factor is associated with age-related decline in hippocampal volume. J. Neurosci. 2010, 30, 5368–5375. [Google Scholar] [CrossRef] [Green Version]
- Chupel, M.U.; Minuzzi, L.G.; Furtado, G.E.; Santos, M.L.; Ferreira, J.P.; Filaire, E.; Teixeira, A.M. Taurine supplementation reduces myeloperoxidase and matrix-metalloproteinase-9 levels and improves the effects of exercise in cognition and physical fitness in older women. Amino Acids 2021, 53, 333–345. [Google Scholar] [CrossRef]
- Nadarajah, V.D.; van Putten, M.; Chaouch, A.; Garrood, P.; Straub, V.; Lochmüller, H.; Ginjaar, H.B.; Aartsma-Rus, A.M.; van Ommen, G.J.; den Dunnen, J.T.; et al. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in Duchenne muscular dystrophy (DMD). Neuromuscul. Disord. 2011, 21, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Chupel, M.U.; Minuzzi, L.G.; Furtado, G.; Santos, M.L.; Hogervorst, E.; Filaire, E.; Teixeira, A.M. Exercise and taurine in inflammation, cognition, and peripheral markers of blood-brain barrier integrity in older women. Appl. Physiol. Nutr. Metab. 2018, 43, 733–741. [Google Scholar] [CrossRef] [Green Version]
- Nee, L.E.; McMorrow, T.; Campbell, E.; Slattery, C.; Ryan, M.P. TNF-alpha and IL-1beta-mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int. 2004, 66, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.C.; Chiu, C.C.; Chen, L.J.; Hsu, T.C.; Tzang, B.S. Effects of taurine on striatal dopamine transporter expression and dopamine uptake in SHR rats. Behav. Brain Res. 2018, 348, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ripps, H.; Shen, W. Review: Taurine: A “very essential” amino acid. Mol. Vis. 2012, 18, 2673–2686. [Google Scholar]
- Chen, C.; Xia, S.; He, J.; Lu, G.; Xie, Z.; Han, H. Roles of taurine in cognitive function of physiology, pathologies and toxication. Life Sci. 2019, 231, 116584. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.S.; Jun, S.B.; Lovinger, D.; Kim, H.Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, V.K.; Huang, B.X.; Kim, H.Y. Effects of docosahexaenoic acid on mouse brain synaptic plasma membrane proteome analyzed by mass spectrometry and (16)O/(18)O labeling. J. Proteome Res. 2011, 10, 5472–5480. [Google Scholar] [CrossRef] [Green Version]
- Yurko-Mauro, K.; McCarthy, D.; Rom, D.; Nelson, E.B.; Ryan, A.S.; Blackwell, A.; Salem, N., Jr.; Stedman, M. Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimer’s Dement. 2010, 6, 456–464. [Google Scholar] [CrossRef]
- Freeman, H.E.; Klein, R.E.; Townsend, J.W.; Lechtig, A. Nutrition and cognitive development among rural Guatemalan children. Am. J. Public Health 1980, 70, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Johnston, F.E.; Low, S.M.; de Baessa, Y.; MacVean, R.B. Interaction of nutritional and socioeconomic status as determinants of cognitive development in disadvantaged urban Guatemalan children. Am. J. Phys. Anthropol. 1987, 73, 501–506. [Google Scholar] [CrossRef]
- Naveed, S.; Lakka, T.; Haapala, E.A. An Overview on the Associations between Health Behaviors and Brain Health in Children and Adolescents with Special Reference to Diet Quality. Int. J. Environ. Res. Public Health 2020, 17, 953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanjiang, G.; Kleinman, R.E. Nutrition and performance in children. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 342–347. [Google Scholar] [CrossRef]
- Grantham-McGregor, S. A review of studies of the effect of severe malnutrition on mental development. J. Nutr. 1995, 125, 2233S–2238S. [Google Scholar] [CrossRef] [PubMed]
- Black, M.M. Impact of Nutrition on Growth, Brain, and Cognition. Nestle Nutr. Inst. Workshop Ser. 2018, 89, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Power, S.E.; O’Connor, E.M.; Ross, R.P.; Stanton, C.; O’Toole, P.W.; Fitzgerald, G.F.; Jeffery, I.B. Dietary glycaemic load associated with cognitive performance in elderly subjects. Eur. J. Nutr. 2015, 54, 557–568. [Google Scholar] [CrossRef]
- Kang, J.H.; Ascherio, A.; Grodstein, F. Fruit and vegetable consumption and cognitive decline in aging women. Ann. Neurol. 2005, 57, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Keys, A.; Menotti, A.; Karvonen, M.J.; Aravanis, C.; Blackburn, H.; Buzina, R.; Djordjevic, B.S.; Dontas, A.S.; Fidanza, F.; Keys, M.H.; et al. The diet and 15-year death rate in the seven countries study. Am. J. Epidemiol. 1986, 124, 903–915. [Google Scholar] [CrossRef]
- Wu, L.; Sun, D. Adherence to Mediterranean diet and risk of developing cognitive disorders: An updated systematic review and meta-analysis of prospective cohort studies. Sci. Rep. 2017, 7, 41317. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, C.A.; Yannakoulia, M.; Kosmidis, M.H.; Dardiotis, E.; Hadjigeorgiou, G.M.; Sakka, P.; Arampatzi, X.; Bougea, A.; Labropoulos, I.; Scarmeas, N. Mediterranean diet and cognitive health: Initial results from the Hellenic Longitudinal Investigation of Ageing and Diet. PLoS ONE 2017, 12, e0182048. [Google Scholar] [CrossRef] [Green Version]
- Weiser, M.J.; Butt, C.M.; Mohajeri, M.H. Docosahexaenoic Acid and Cognition throughout the Lifespan. Nutrients 2016, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Swanson, D.; Block, R.; Mousa, S.A. Omega-3 fatty acids EPA and DHA: Health benefits throughout life. Adv. Nutr. 2012, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Macaron, T.; Giudici, K.V.; Bowman, G.L.; Sinclair, A.; Stephan, E.; Vellas, B.; de Souto Barreto, P. Associations of Omega-3 fatty acids with brain morphology and volume in cognitively healthy older adults: A narrative review. Ageing Res. Rev. 2021, 67, 101300. [Google Scholar] [CrossRef] [PubMed]
- Sinn, N.; Milte, C.M.; Street, S.J.; Buckley, J.D.; Coates, A.M.; Petkov, J.; Howe, P.R. Effects of n-3 fatty acids, EPA v. DHA, on depressive symptoms, quality of life, memory and executive function in older adults with mild cognitive impairment: A 6-month randomised controlled trial. Br. J. Nutr. 2012, 107, 1682–1693. [Google Scholar] [CrossRef] [Green Version]
- Marti Del Moral, A.; Fortique, F. Omega-3 fatty acids and cognitive decline: A systematic review. Nutr. Hosp. 2019, 36, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Bozzatello, P.; Rocca, P.; Mantelli, E.; Bellino, S. Polyunsaturated Fatty Acids: What is Their Role in Treatment of Psychiatric Disorders? Int. J. Mol. Sci. 2019, 20, 5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, N.; Werner, S.; Grimm, A.; Eckert, A. Dietary Mitophagy Enhancer: A Strategy for Healthy Brain Aging? Antioxidants 2020, 9, 932. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural. Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Munshi, S.; Banerjee, K.; Thakurta, I.G.; Sinha, M.; Bagh, M.B. Mitochondrial Dysfunction during Brain Aging: Role of Oxidative Stress and Modulation by Antioxidant Supplementation. Aging Dis. 2011, 2, 242–256. [Google Scholar] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Head, E. Oxidative damage and cognitive dysfunction: Antioxidant treatments to promote healthy brain aging. Neurochem. Res. 2009, 34, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Neely, M.D.; Quinn, J.F.; Beal, M.F.; Markesbery, W.R.; Roberts, L.J.; Morrow, J.D. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic. Biol. Med. 2002, 33, 620–626. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Bennett, J.P., Jr. An evaluation of the role of mitochondria in neurodegenerative diseases: Mitochondrial mutations and oxidative pathology, protective nuclear responses, and cell death in neurodegeneration. Brain Res. Brain Res. Rev. 1999, 29, 1–25. [Google Scholar] [CrossRef]
- Percario, S.; da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; de Nazare Araujo Moreira, T.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxid. Med. Cell. Longev. 2020, 2020, 2360872. [Google Scholar] [CrossRef]
- Dowling, A.L.; Head, E. Antioxidants in the canine model of human aging. Biochim. Biophys. Acta 2012, 1822, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milgram, N.W.; Head, E.; Muggenburg, B.; Holowachuk, D.; Murphey, H.; Estrada, J.; Ikeda-Douglas, C.J.; Zicker, S.C.; Cotman, C.W. Landmark discrimination learning in the dog: Effects of age, an antioxidant fortified food, and cognitive strategy. Neurosci. Biobehav. Rev. 2002, 26, 679–695. [Google Scholar] [CrossRef]
- Alam, J. Vitamins: A nutritional intervention to modulate the Alzheimer’s disease progression. Nutr. Neurosci. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pepersack, T.; Garbusinski, J.; Robberecht, J.; Beyer, I.; Willems, D.; Fuss, M. Clinical relevance of thiamine status amongst hospitalized elderly patients. Gerontology 1999, 45, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Eussen, S.J.; de Groot, L.C.; Joosten, L.W.; Bloo, R.J.; Clarke, R.; Ueland, P.M.; Schneede, J.; Blom, H.J.; Hoefnagels, W.H.; van Staveren, W.A. Effect of oral vitamin B-12 with or without folic acid on cognitive function in older people with mild vitamin B-12 deficiency: A randomized, placebo-controlled trial. Am. J. Clin. Nutr. 2006, 84, 361–370. [Google Scholar] [CrossRef]
- Kwok, T.; Lee, J.; Ma, R.C.; Wong, S.Y.; Kung, K.; Lam, A.; Ho, C.S.; Lee, V.; Harrison, J.; Lam, L. A randomized placebo controlled trial of vitamin B12 supplementation to prevent cognitive decline in older diabetic people with borderline low serum vitamin B12. Clin. Nutr. 2017, 36, 1509–1515. [Google Scholar] [CrossRef]
- Moore, E.; Mander, A.; Ames, D.; Carne, R.; Sanders, K.; Watters, D. Cognitive impairment and vitamin B12: A review. Int. Psychogeriatr 2012, 24, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Gale, C.R.; Martyn, C.N.; Cooper, C. Cognitive impairment and mortality in a cohort of elderly people. BMJ 1996, 312, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Fata, G.; Weber, P.; Mohajeri, M.H. Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 2014, 6, 5453–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillenbaum, G.G.; Kuchibhatla, M.N.; Hanlon, J.T.; Artz, M.B.; Pieper, C.F.; Schmader, K.E.; Dysken, M.W.; Gray, S.L. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann. Pharmacother. 2005, 39, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Rutjes, A.W.; Denton, D.A.; Di Nisio, M.; Chong, L.Y.; Abraham, R.P.; Al-Assaf, A.S.; Anderson, J.L.; Malik, M.A.; Vernooij, R.W.; Martinez, G.; et al. Vitamin and mineral supplementation for maintaining cognitive function in cognitively healthy people in mid and late life. Cochrane Database Syst. Rev. 2018, 12, CD011906. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Vinciguerra, F.; Graziano, M.; Hagnas, M.; Frittitta, L.; Tumminia, A. Influence of the Mediterranean and Ketogenic Diets on Cognitive Status and Decline: A Narrative Review. Nutrients 2020, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49 (Suppl. 8), 3–5. [Google Scholar] [CrossRef]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ulamek-Koziol, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef]
- Liu, Y.M.; Wang, H.S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Ohnuma, T.; Toda, A.; Kimoto, A.; Takebayashi, Y.; Higashiyama, R.; Tagata, Y.; Ito, M.; Ota, T.; Shibata, N.; Arai, H. Benefits of use, and tolerance of, medium-chain triglyceride medical food in the management of Japanese patients with Alzheimer’s disease: A prospective, open-label pilot study. Clin. Interv. Aging 2016, 11, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Sastre, J.; Millan, A.; Garcia de la Asuncion, J.; Pla, R.; Juan, G.; Pallardo; O’Connor, E.; Martin, J.A.; Droy-Lefaix, M.T.; Vina, J. A Ginkgo biloba extract (EGb 761) prevents mitochondrial aging by protecting against oxidative stress. Free Radic. Biol. Med. 1998, 24, 298–304. [Google Scholar] [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef] [Green Version]
- Braidy, N.; Essa, M.M.; Poljak, A.; Selvaraju, S.; Al-Adawi, S.; Manivasagm, T.; Thenmozhi, A.J.; Ooi, L.; Sachdev, P.; Guillemin, G.J. Consumption of pomegranates improves synaptic function in a transgenic mice model of Alzheimer’s disease. Oncotarget 2016, 7, 64589–64604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, M.R.; Nabavi, S.M.; Braidy, N.; Setzer, W.N.; Ahmed, T.; Nabavi, S.F. Quercetin and the mitochondria: A mechanistic view. Biotechnol. Adv. 2016, 34, 532–549. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Barnes, L.L.; Bennett, D.A.; Aggarwal, N.T. MIND diet slows cognitive decline with aging. Alzheimer’s Dement. 2015, 11, 1015–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaili, M. Effect of diet and nutrients on molecular mechanism of gene expression mediated by nuclear receptor and epigenetic moduluation. Open Nutraceuticals J. 2013, 6, 27–34. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaliszewska, A.; Allison, J.; Martini, M.; Arias, N. The Interaction of Diet and Mitochondrial Dysfunction in Aging and Cognition. Int. J. Mol. Sci. 2021, 22, 3574. https://doi.org/10.3390/ijms22073574
Kaliszewska A, Allison J, Martini M, Arias N. The Interaction of Diet and Mitochondrial Dysfunction in Aging and Cognition. International Journal of Molecular Sciences. 2021; 22(7):3574. https://doi.org/10.3390/ijms22073574
Chicago/Turabian StyleKaliszewska, Aleksandra, Joseph Allison, Matteo Martini, and Natalia Arias. 2021. "The Interaction of Diet and Mitochondrial Dysfunction in Aging and Cognition" International Journal of Molecular Sciences 22, no. 7: 3574. https://doi.org/10.3390/ijms22073574
APA StyleKaliszewska, A., Allison, J., Martini, M., & Arias, N. (2021). The Interaction of Diet and Mitochondrial Dysfunction in Aging and Cognition. International Journal of Molecular Sciences, 22(7), 3574. https://doi.org/10.3390/ijms22073574